
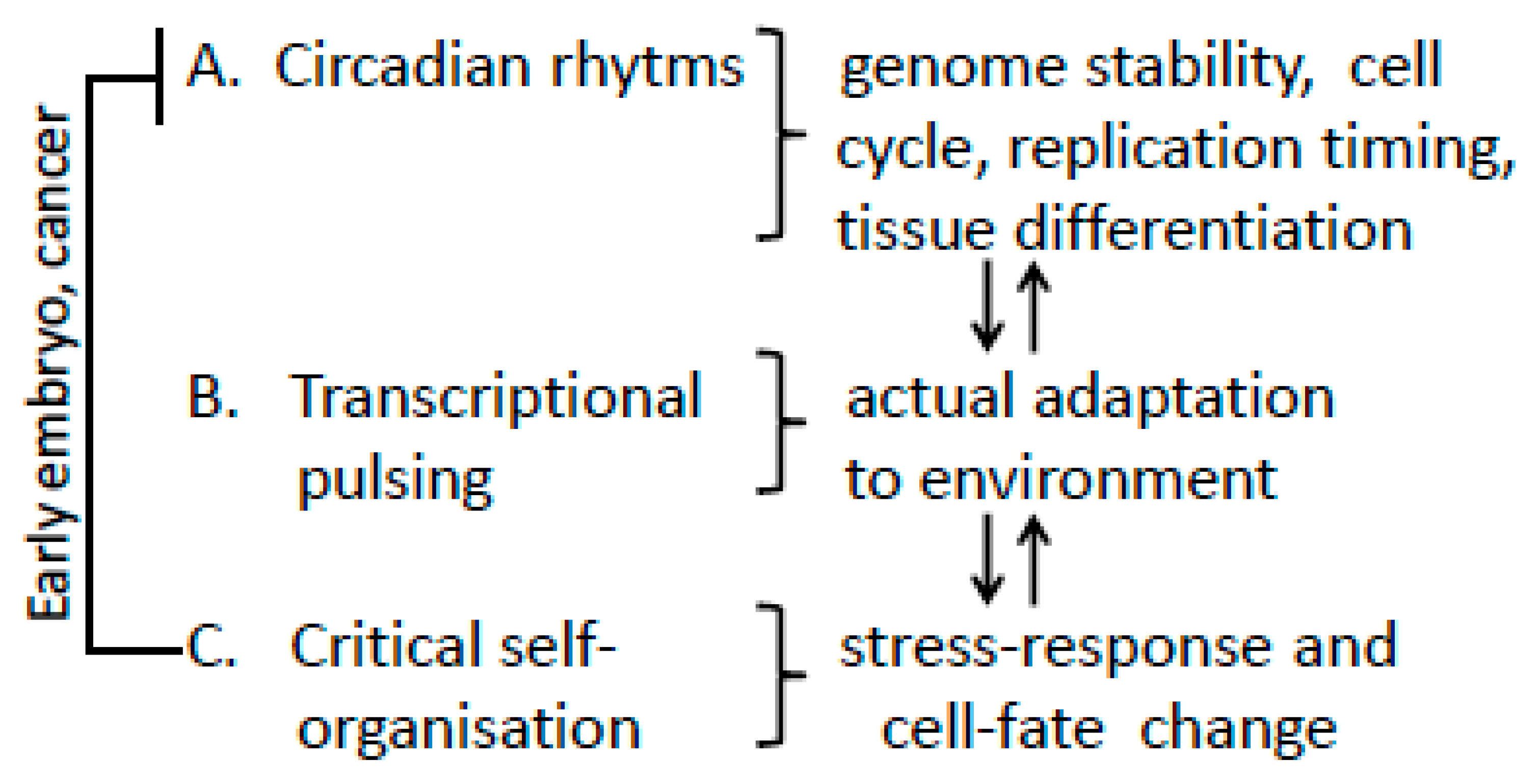
Spatial-Temporal Genome Regulation in Stress-Response and Cell-Fate Change
 , ,
, ,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sessions and Presentations
2.1. The Guardians of Stability Are the Same That Initiate Revolutions: The Oscillatory Principle of Gene Expression Dynamics (Presented by A. Giuliani)
2.1.1. Setting the Scenes
- (a)
- Biological scene (size matters):
- (b)
- Physical scene (sandpile tales):
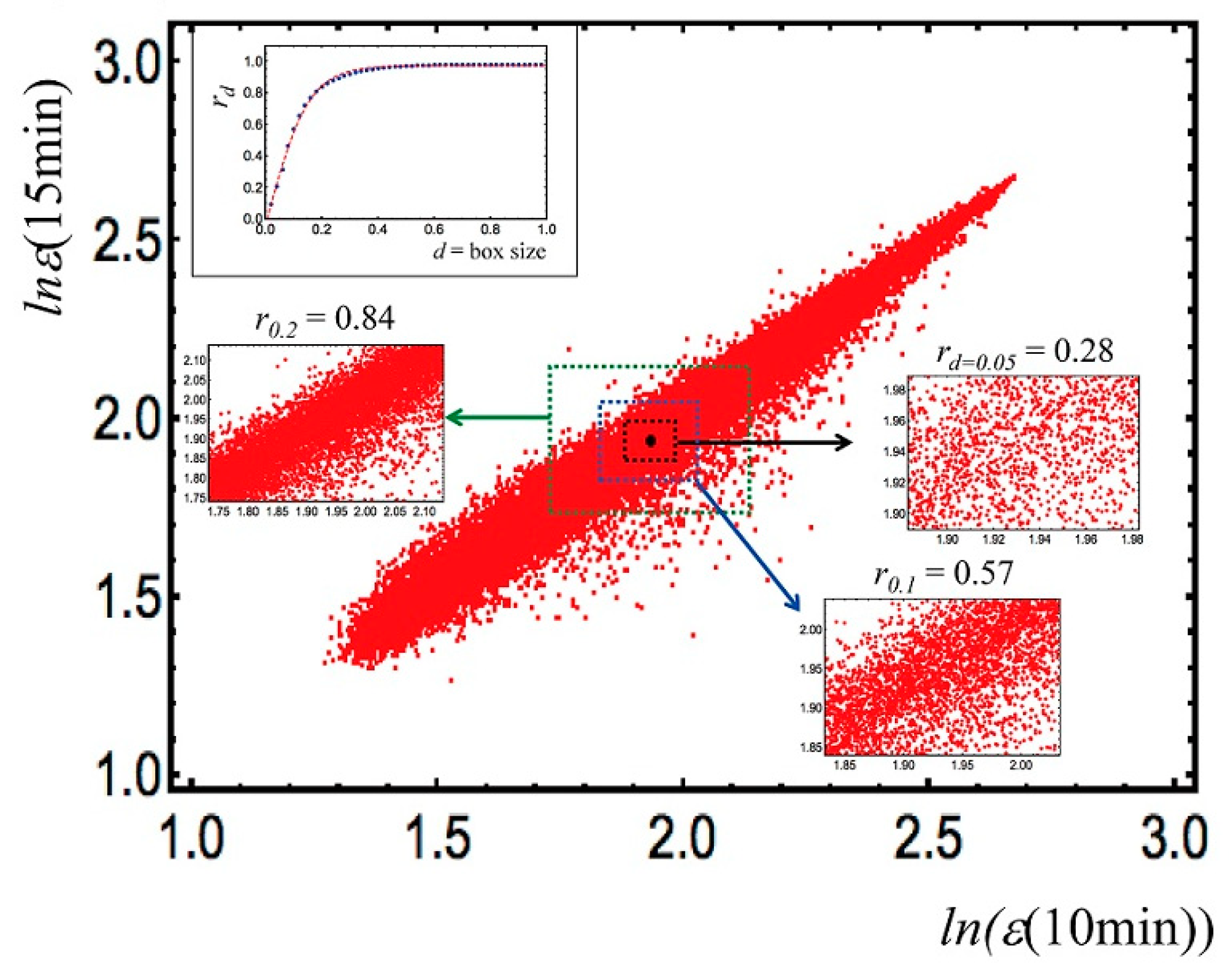
2.1.2. Gene Expression Regulation at the Genomic Scale (Putting SOC at Work)
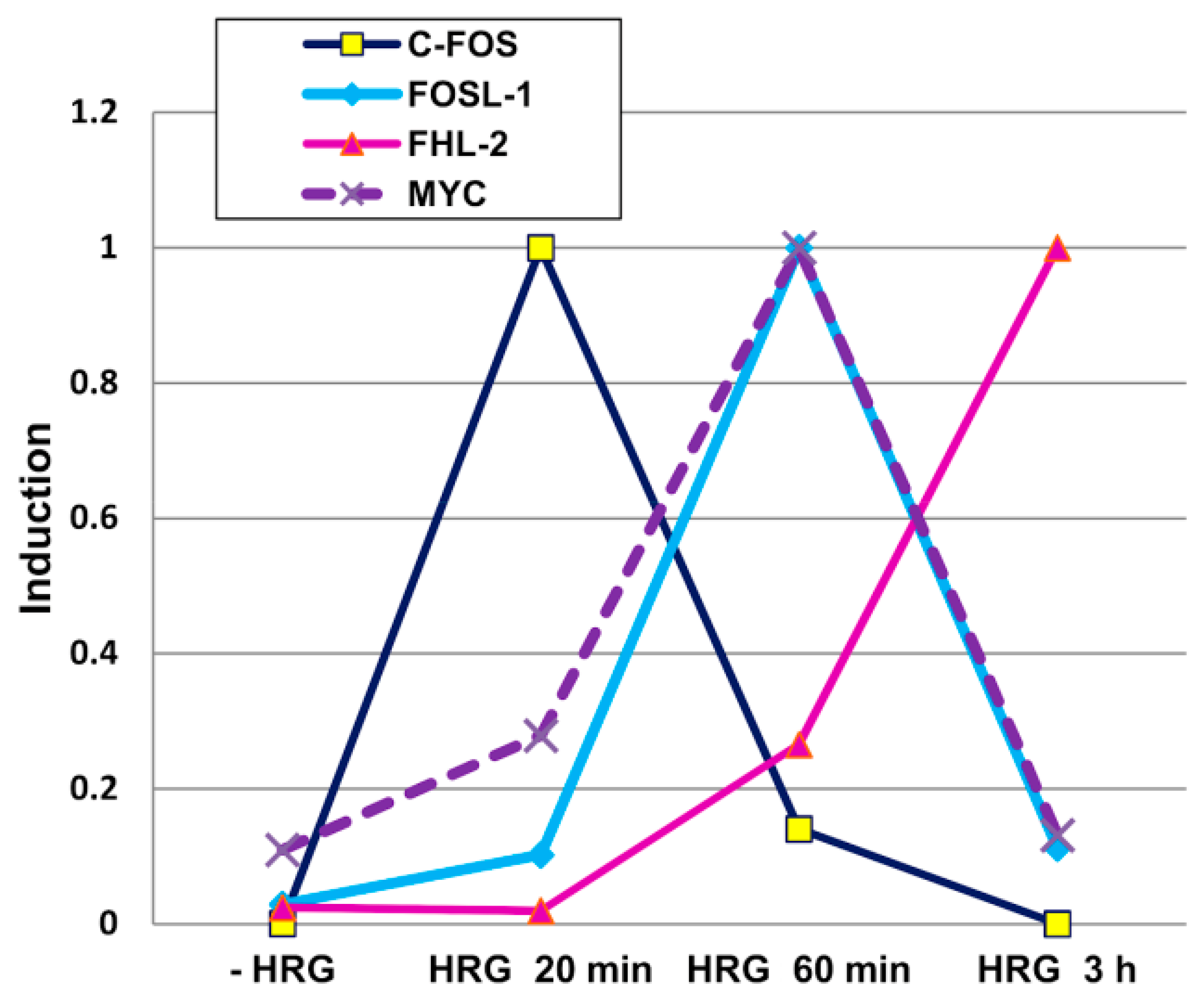
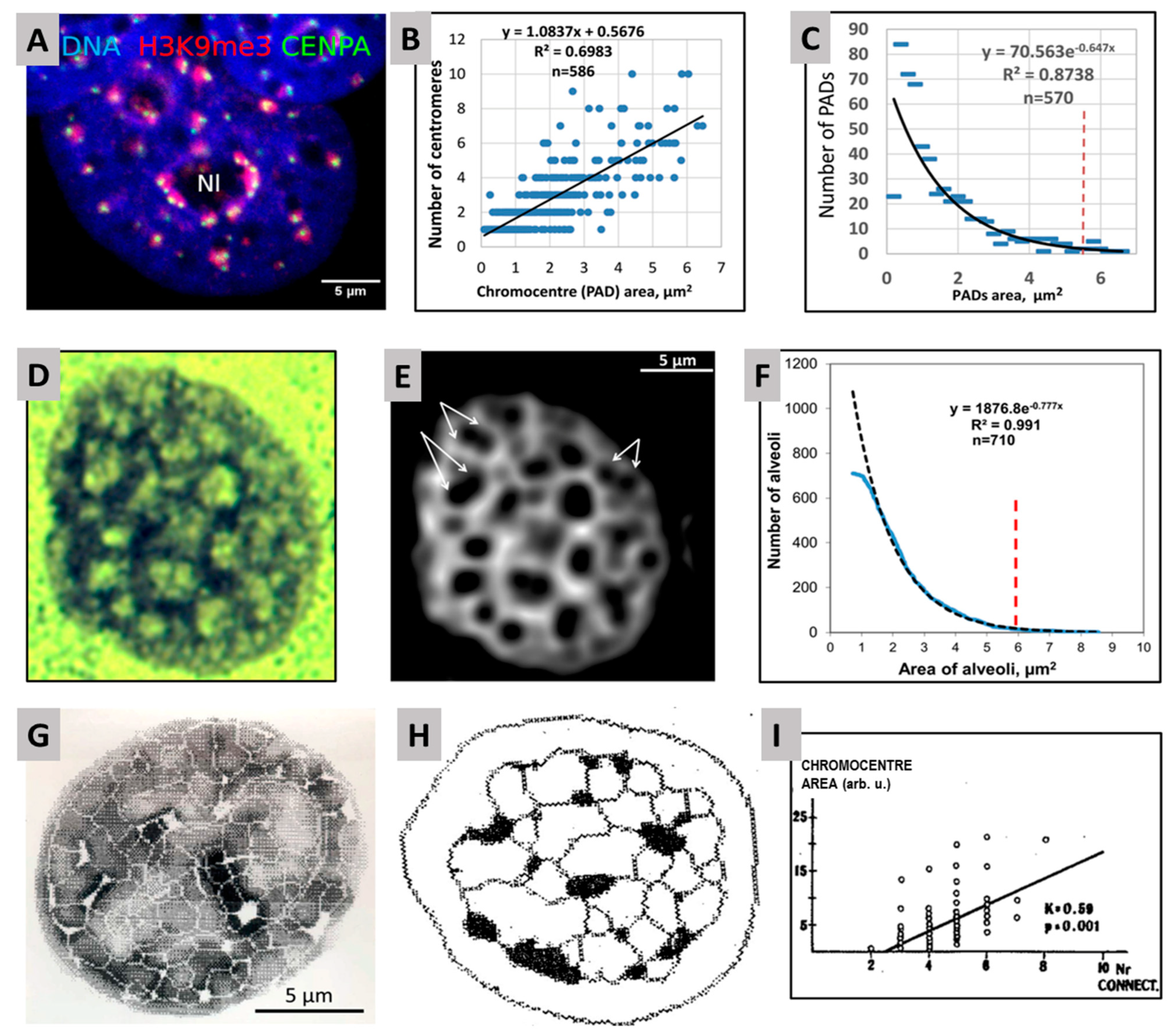
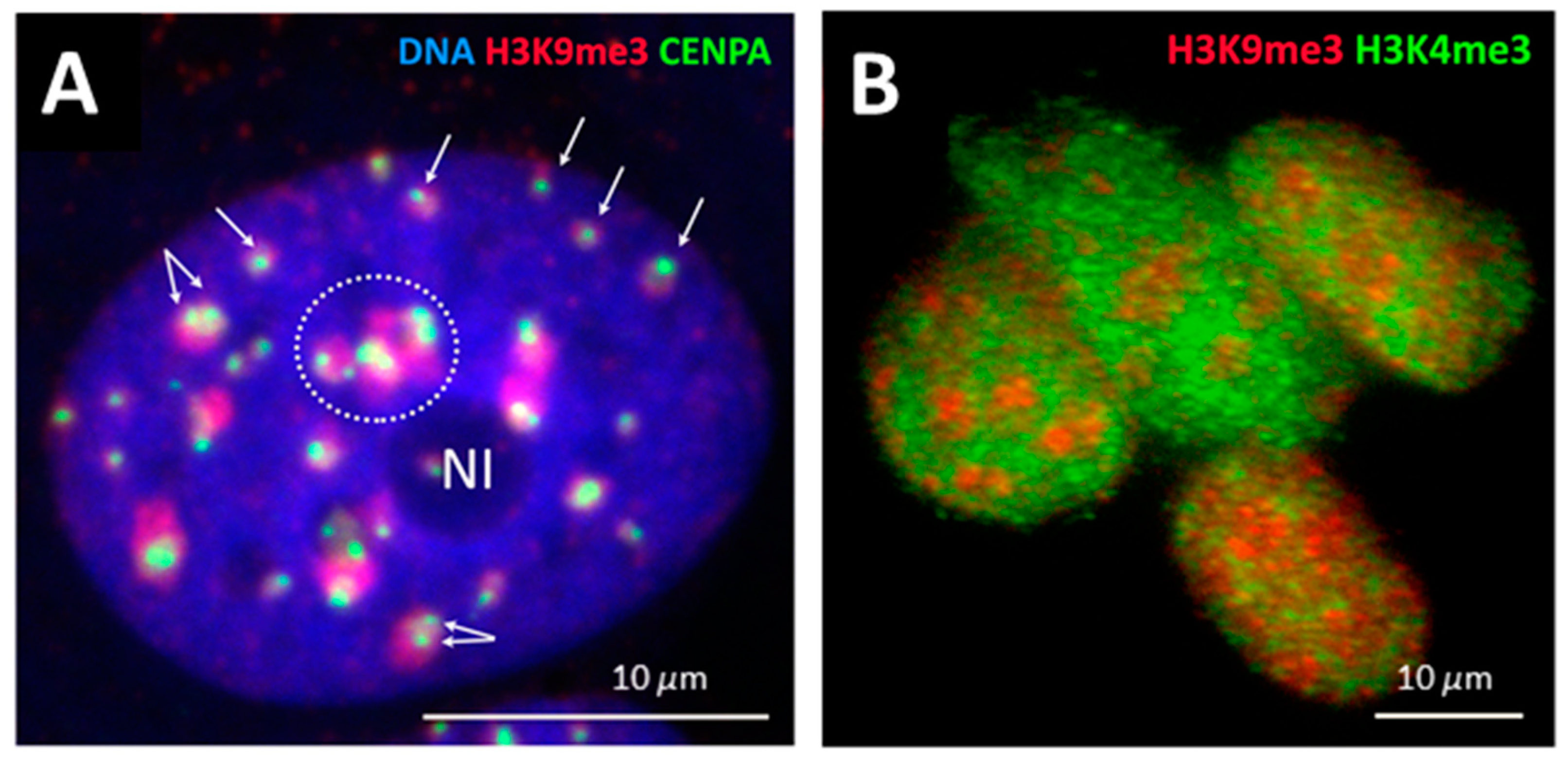
2.2. Genome Regulation by Positional Information in Space and Time (Presented by J. Erenpreisa)
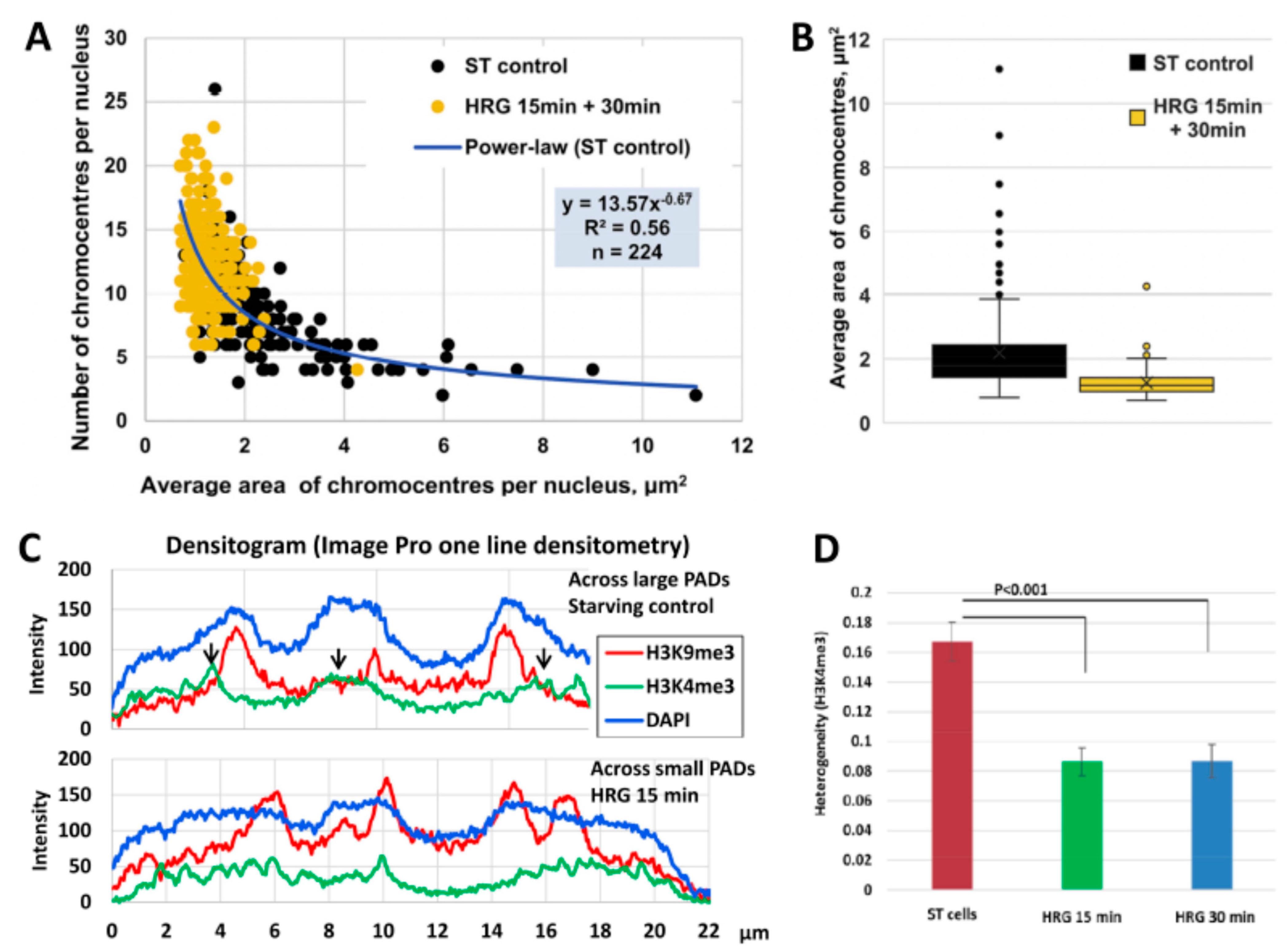
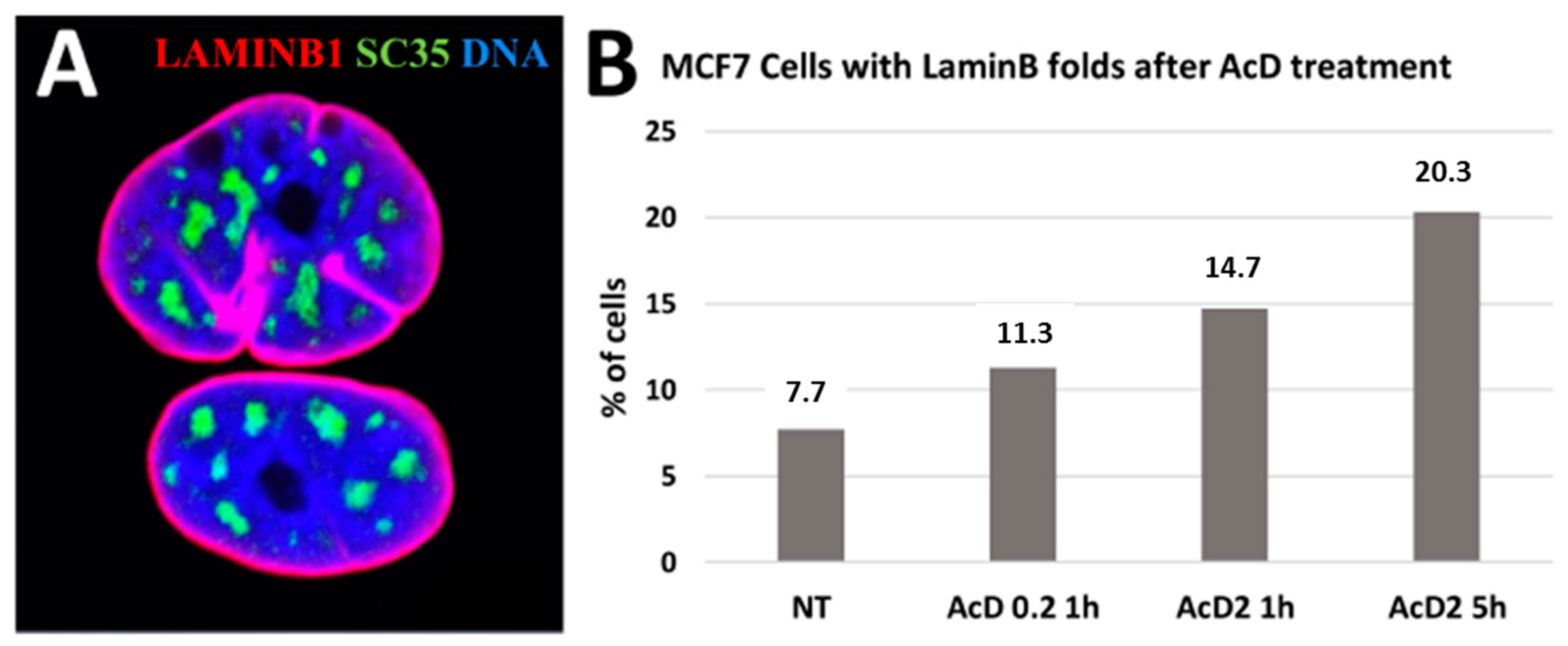
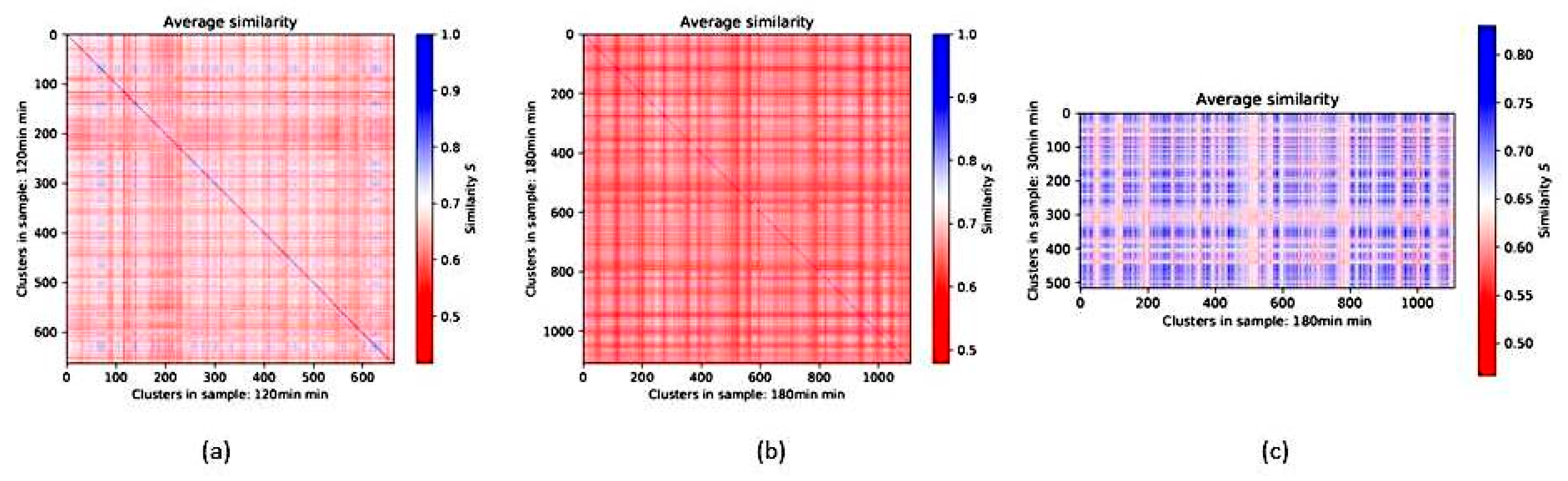
2.3. Scale-Free Organization of Pericentric Chromatin Domains in MCF7 Breast Cancer Chromatin Nuclei and the Impact of HRG Treatment (Contributed by T. Freivalds)
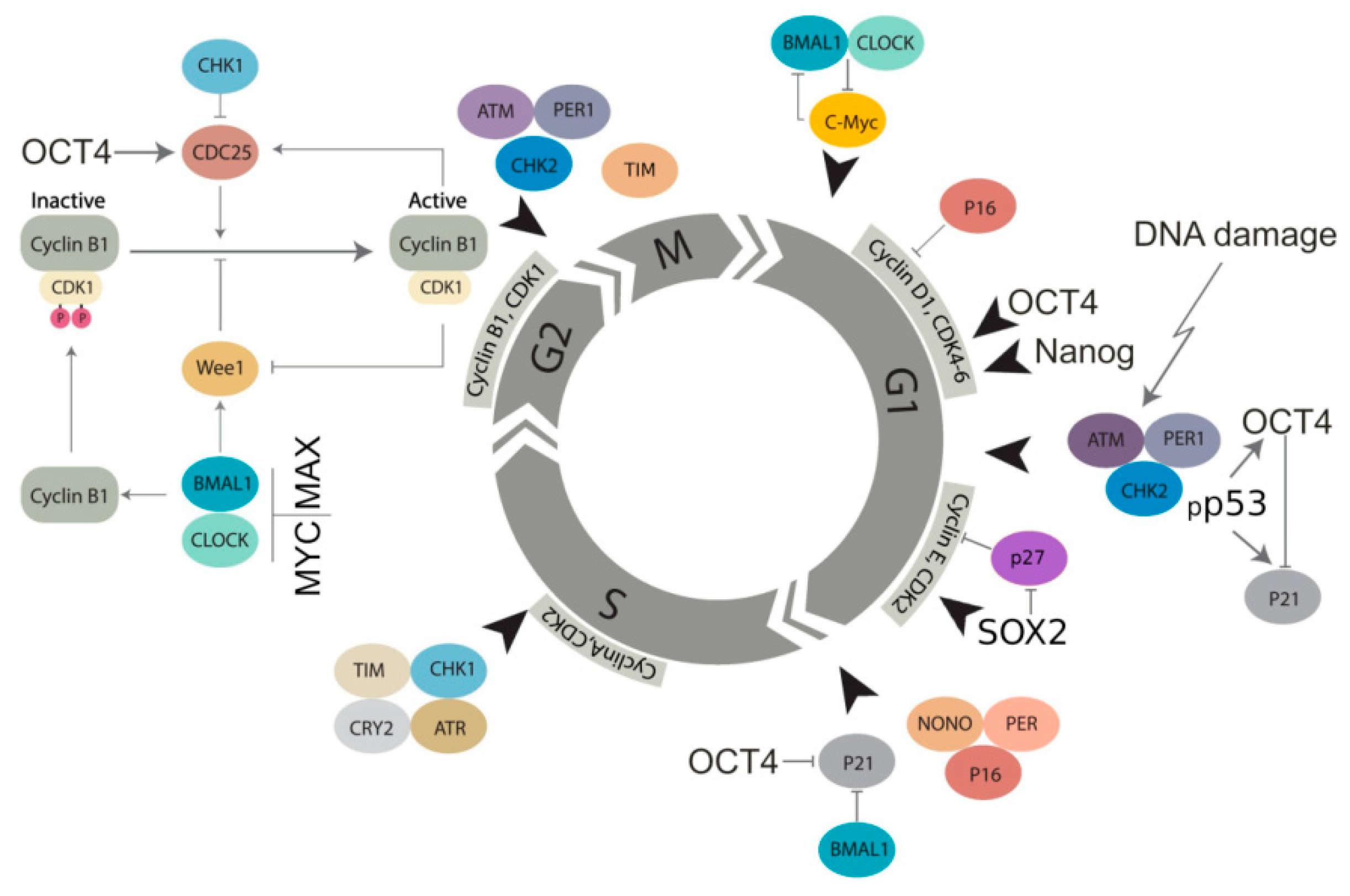
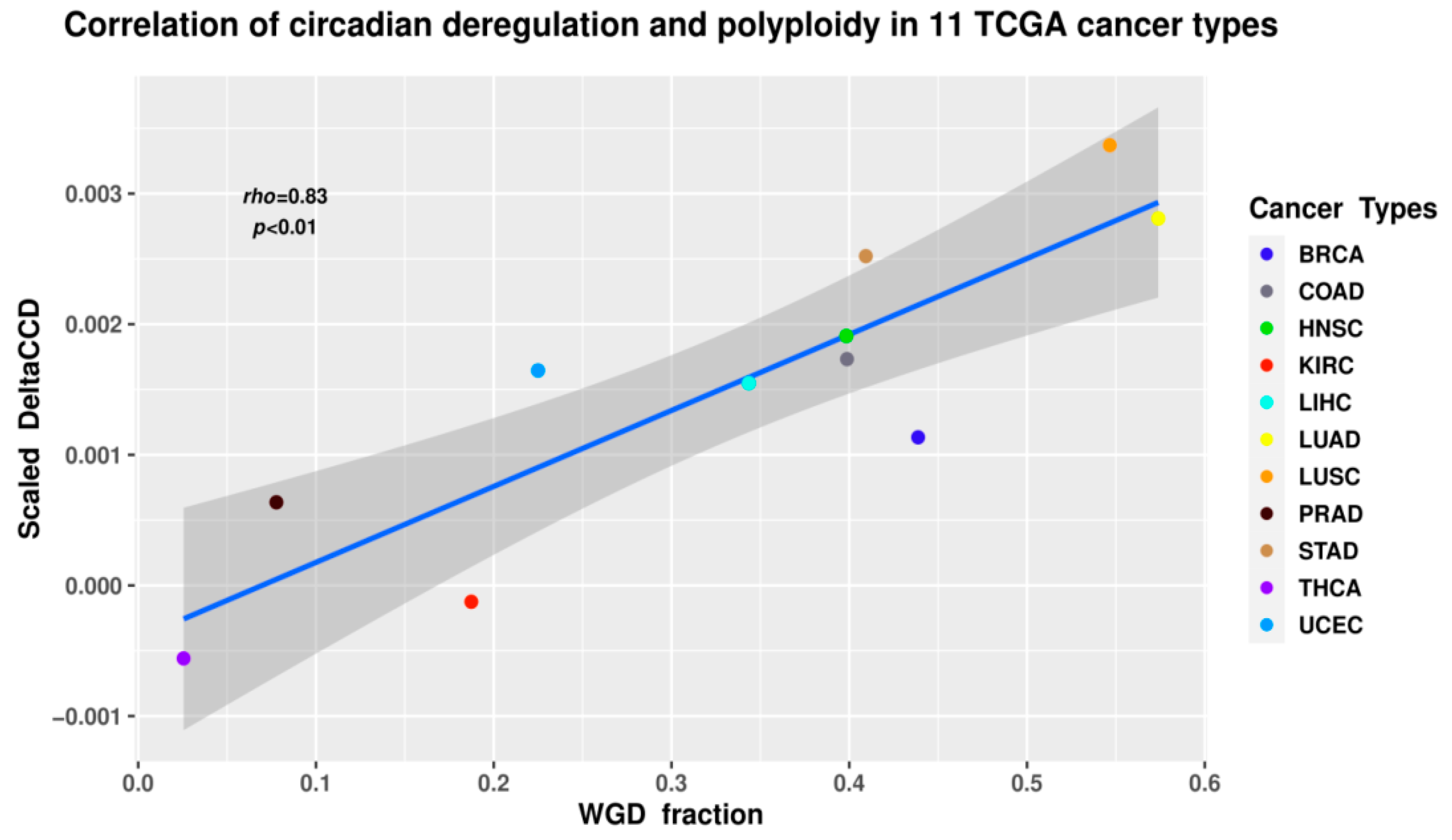
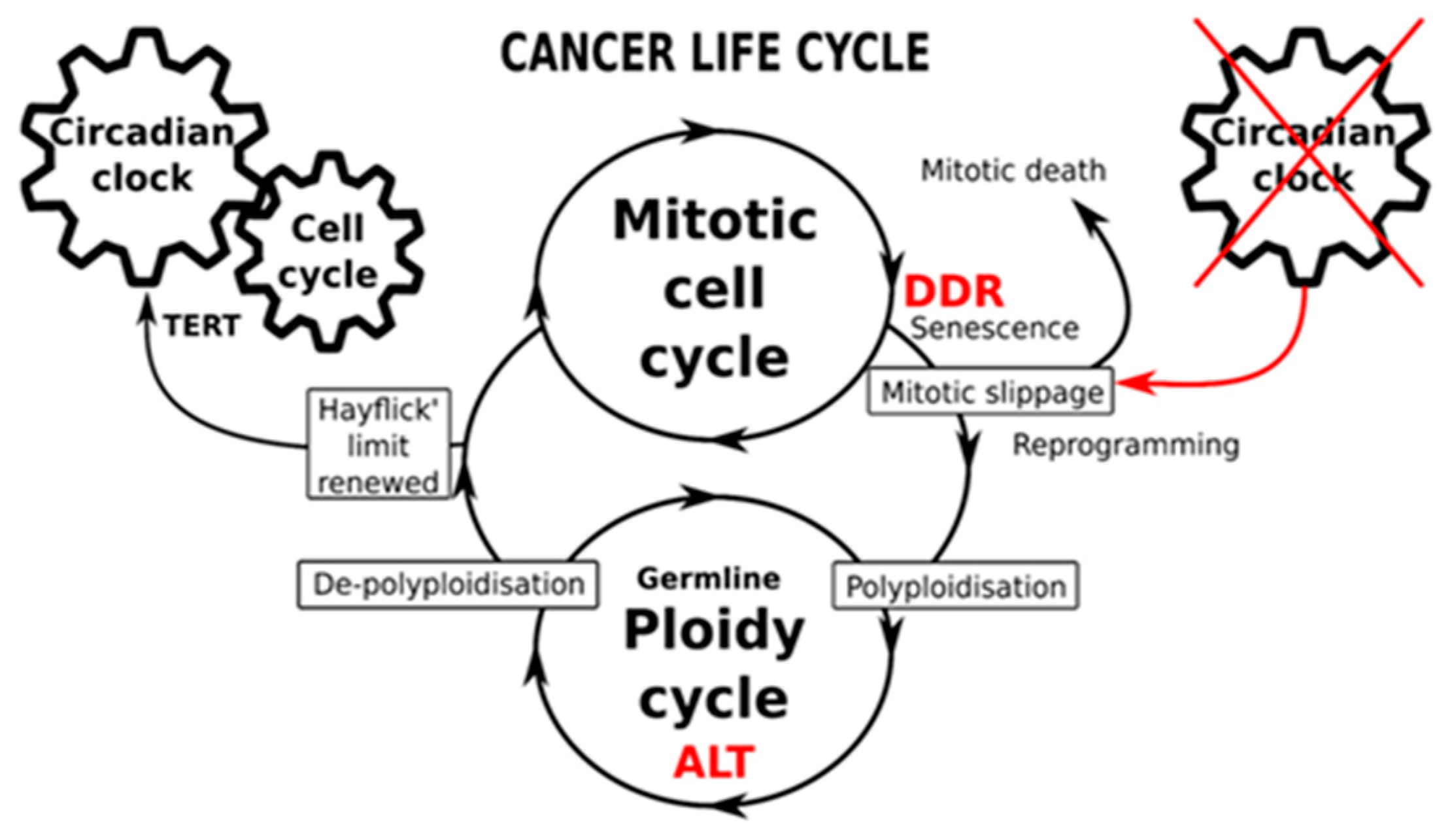
2.4. The Circadian Clock and Cancer (Presented by N.M. Vainshelbaum)
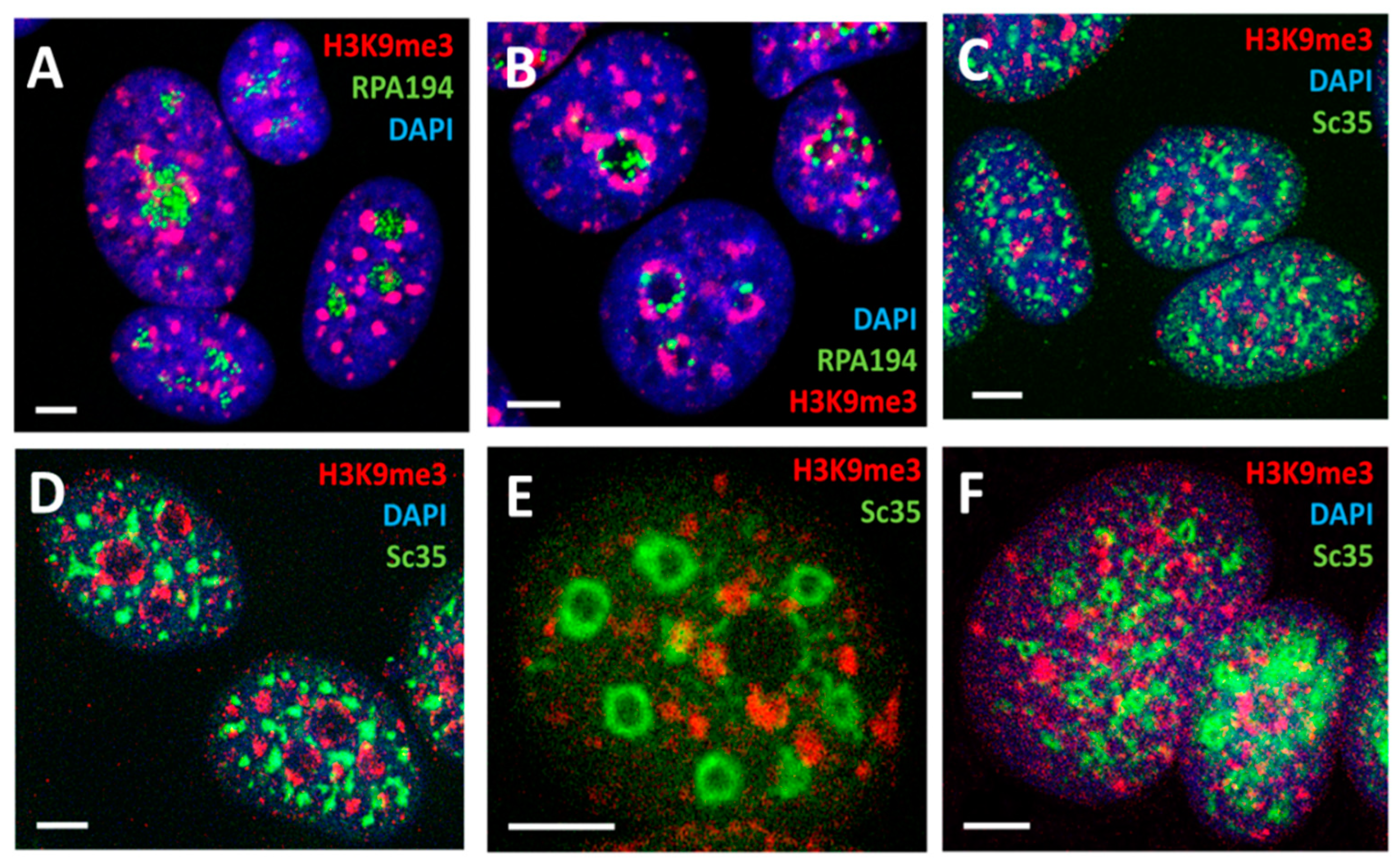
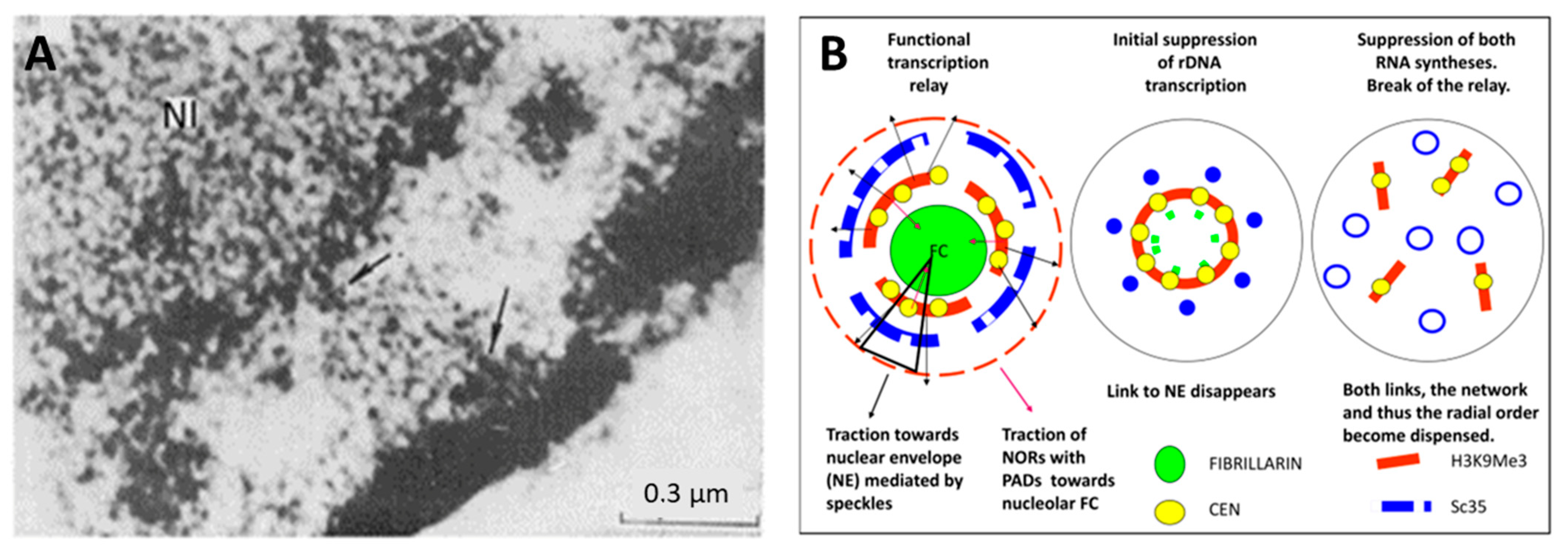
2.5. Spatial Relationship between Ribosomal and mRNA Transcription/Splicing Conveyor, Nuclear Lamin Rigidity, and Actin Cytoskeleton Tension (Presented by K. Salmina)
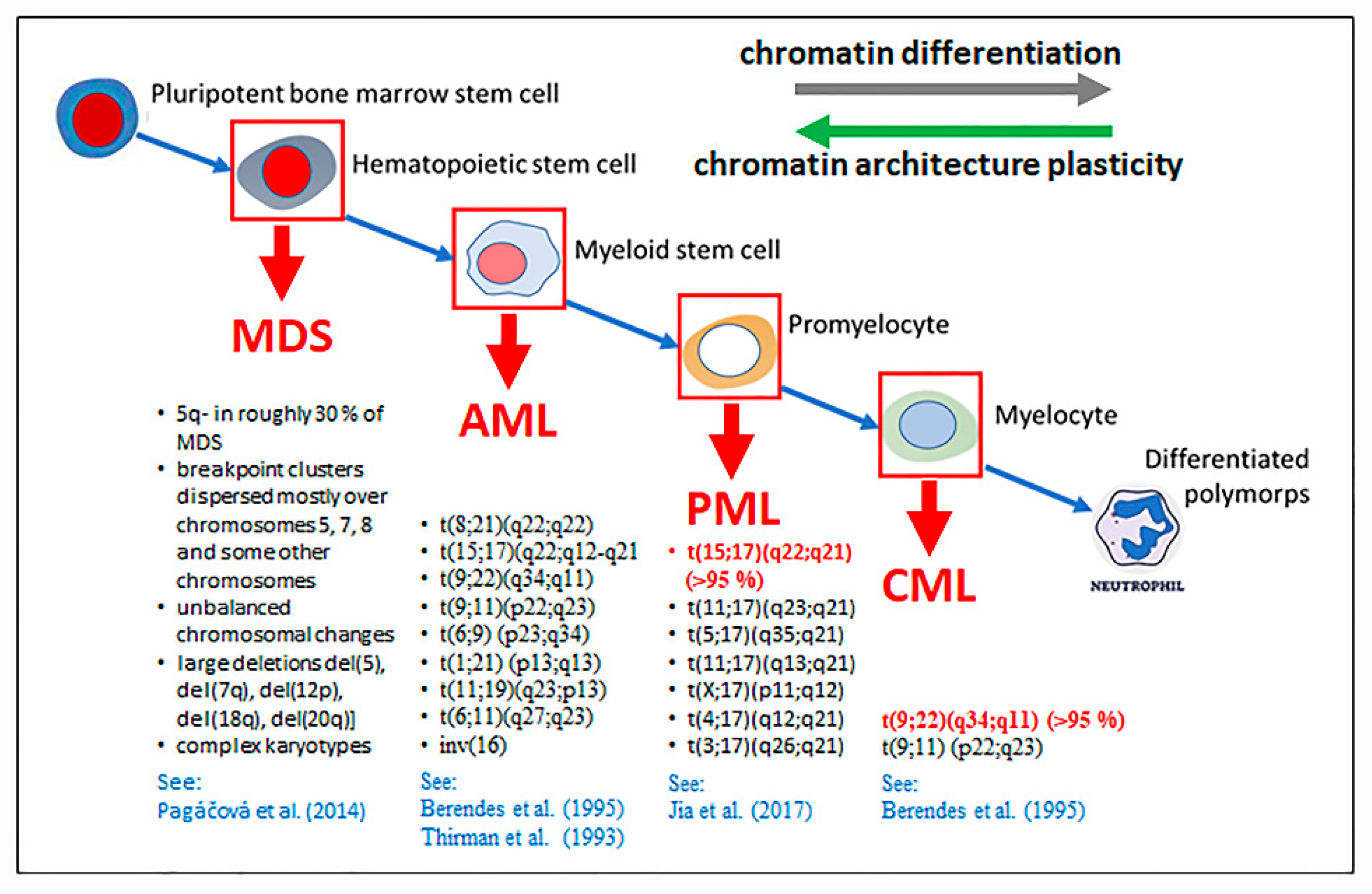
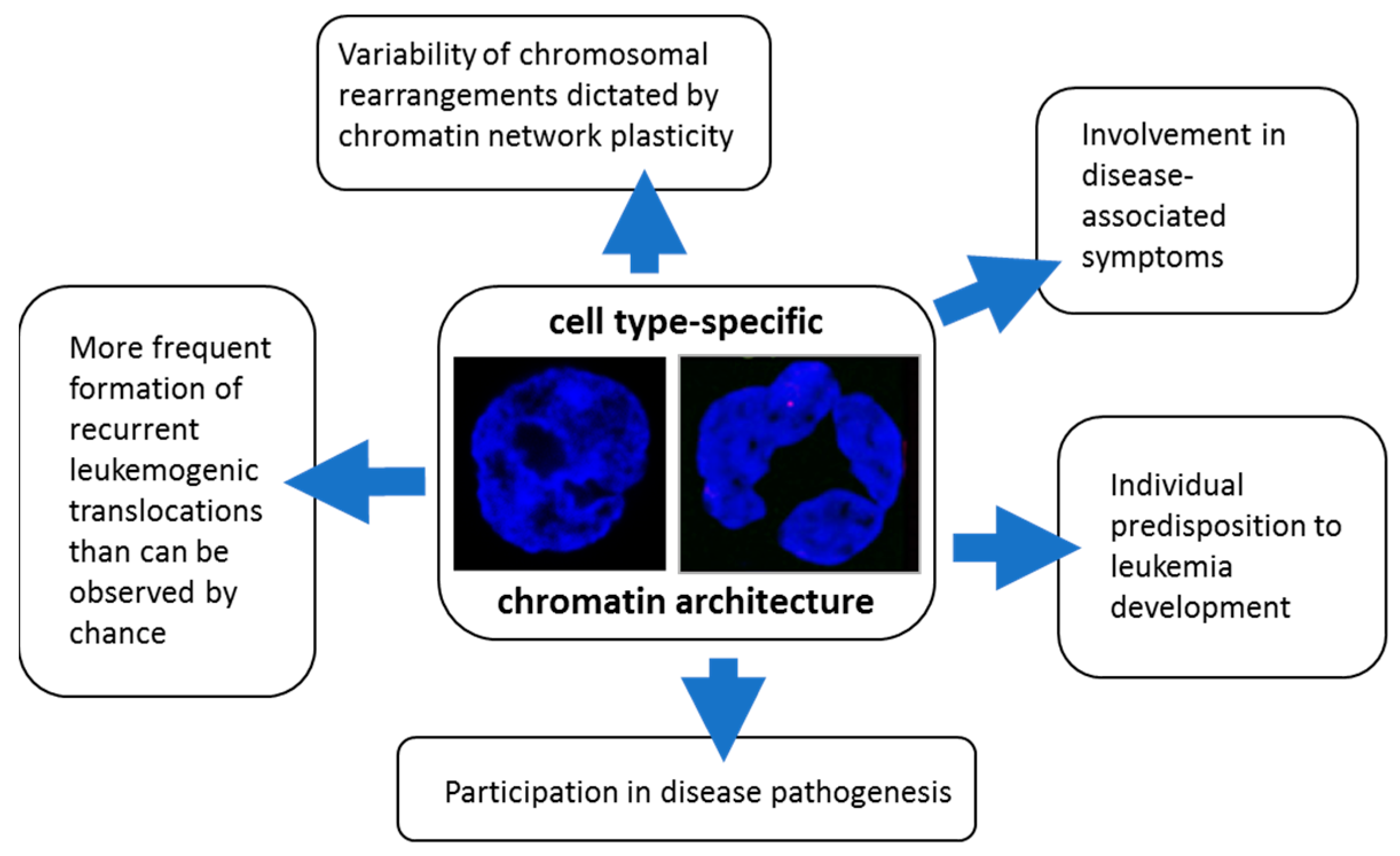
2.6. How Nanoscale Chromatin Architecture and Chromatin Topology within the Cell Nucleus Participates in Cancer Development—An Example of Pathogenesis of Three Different Leukemia Types (Presented by M. Falk)
2.6.1. Chromatin and Cell Nucleus Architecture in Formation of Chromosomal Translocations—General Aspects
2.6.2. Roles of Chromatin Architecture and Its Plasticity in of the Development of Recurrent Chromosomal Aberrations Characteristic for Different Types of Leukemia
2.6.3. New Insights into the Genesis of (Leukemogenic) Translocations at the Nanoresolution Level
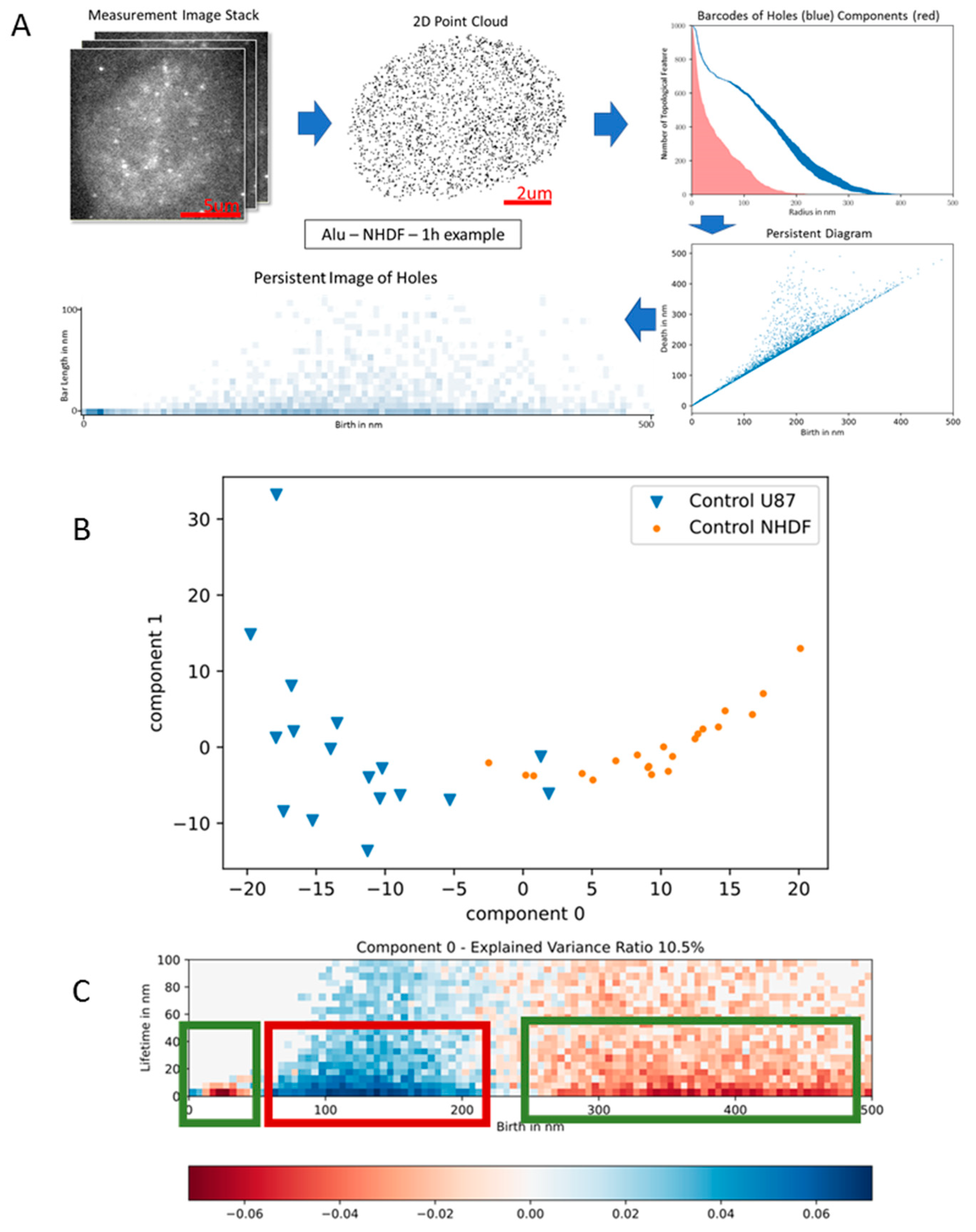
2.7. Schrödinger’s Chromosomal Aperiodic Crystal: A Model for Radiation Response of the Cell Nucleus (Presented by M. Hausmann)
2.8. Significance of Non-Specific Physical Parameters on Genetic Activity through the Transition of the Higher-Order DNA Structure (Presented by K. Yoshikawa)
2.8.1. Discrete Transition of Individual DNA
2.8.2. Inhibition/Promotion of Genetic Activity Accompanied by the Conformational Change of DNA
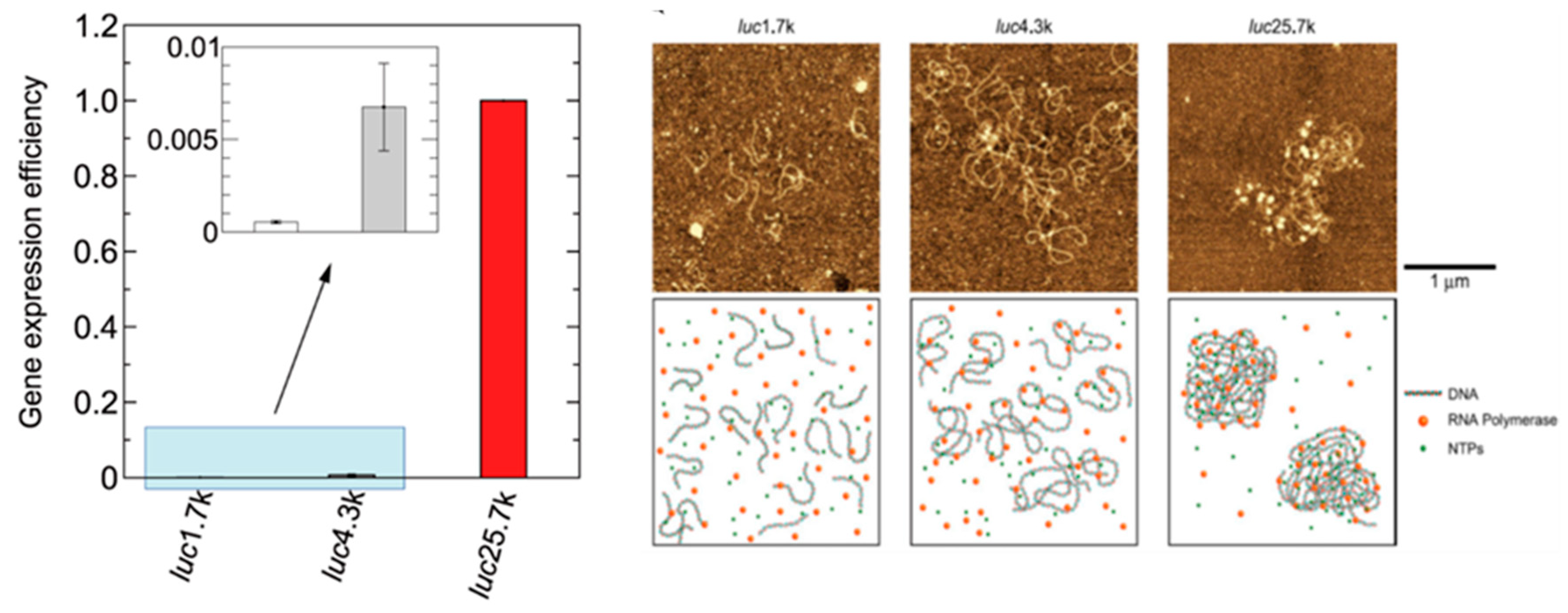
2.8.3. Longer DNA Exhibits Higher Genetic Activity
2.8.4. Phase Segregation in Single DNA
2.8.5. Importance of Aspecific Parameters
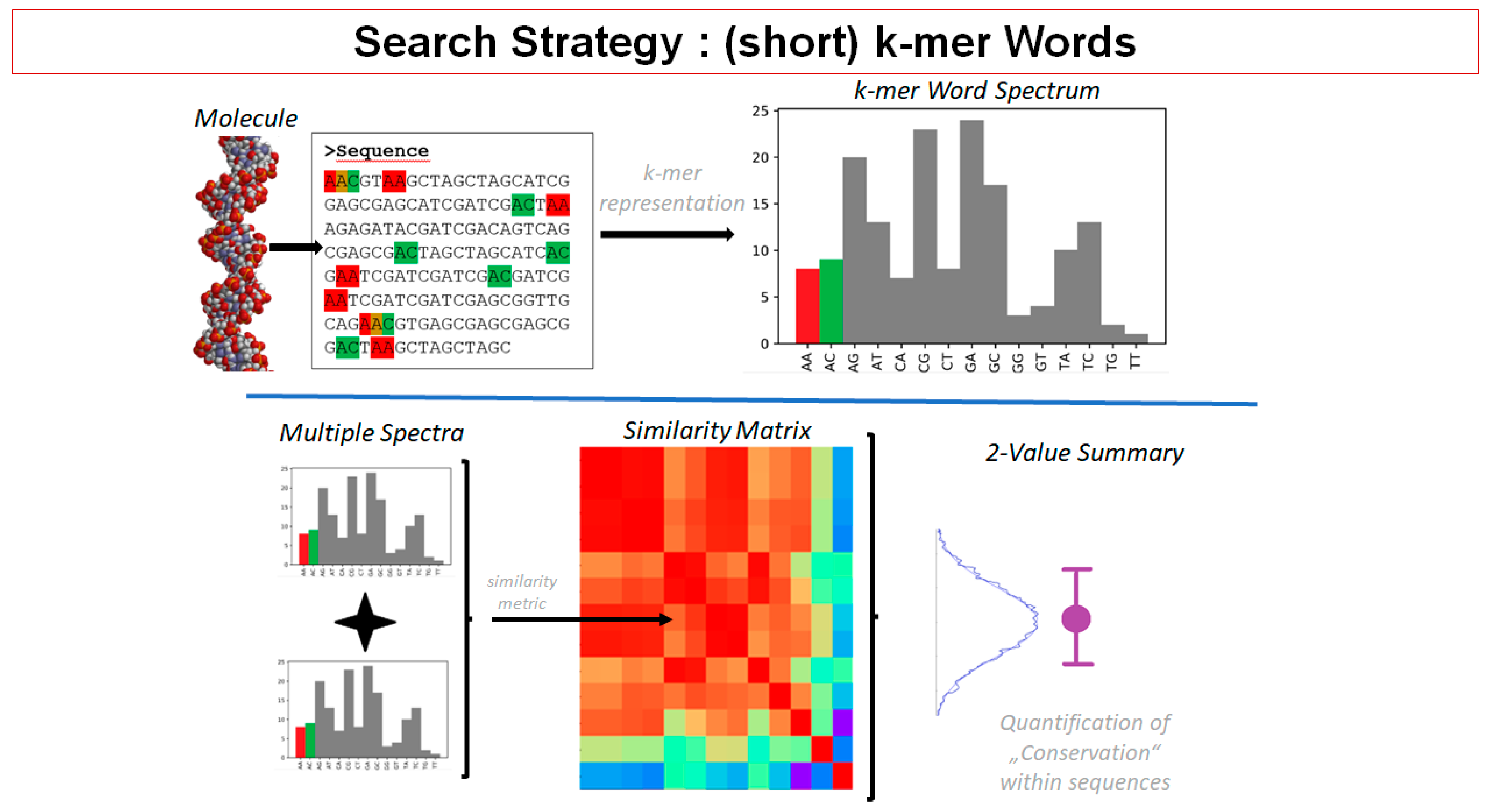
2.9. Insights from Alignment-Free Analyses of DNA Data Bases (Presented by G. Hildenbrand)
- (1)
- Search: Find conserved genomic DNA patterns;
- (2)
- Analysis: Look for association with known functional elements. Look for properties of identified DNA sequences;
- (3)
- Interpretation: Make conclusions on function of found DNA patterns.
- (1)
- Conserved patterns based on k-words and SSTRs are found in genomes;
- (2)
- Strong correlations/anticorrelations with biophysical parameters are found;
- (3)
- Organization of DNA may not only be based on long conserved sequences but also on base composition and length of SSTRs.
3. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vickaryous, M.K.; Hall, B.K. Human Cell Type Diversity, Evolution, Development, and Classification with Special Reference to Cells Derived from the Neural Crest. Biol. Rev. Camb. Philos. Soc. 2006, 81, 425–455. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, M.; Hildenbrand, G.; Pilarczyk, G. Networks and Islands of Genome Nano-Architecture and Their Potential Relevance for Radiation Biology (A Hypothesis and Experimental Verification Hints). In Nuclear, Chromosomal, and Genomic Architecture in Biology and Medicine; Results and Problems in Cell Differentiation; Kloc, M., Kubiak, J.Z., Eds.; Springer: Cham, Switzerland, 2022; Volume 70, pp. 3–34. [Google Scholar]
- Karsenti, E. Self-organization in cell biology: A brief history. Nat. Rev. Mol. Cell Biol. 2008, 9, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Beyond the sequence: Cellular organization of genome function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misteli, T. The self-organizing genome: Principles of genome architecture and function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transistions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Giuliani, A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. Int. J. Mol. Sci. 2019, 21, 240. [Google Scholar] [CrossRef] [Green Version]
- Zwieniecki, M.A.; Holbrook, N.M. Confronting Maxwell’s Demon: Biophysics of Xylem Embolism Repair. Trends Plant Sci. 2009, 14, 530–534. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Giuliani, A.; Hashimoto, M.; Erenpreisa, J.; Yoshikawa, K. Emergent Self-Organized Criticality in Gene Expression Dynamics: Temporal Development of Global Phase Transition Revealed in a Cancer Cell Line. PLoS ONE 2015, 10, e0128565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-Organization of Whole-Gene Expression through Coordinated Chromatin Structural Transition. Biophys. Rev. 2021, 2, 031303. [Google Scholar] [CrossRef]
- Turcotte, D.L.; Rundle, J.B. Self-Organized Complexity in the Physical, Biological, and Social Sciences. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S1), 2463–2465. [Google Scholar] [CrossRef]
- Huang, S. Reconciling Non-Genetic Plasticity with Somatic Evolution in Cancer. Trends Cancer Res. 2021, 7, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.M.W. Conrad Hal Waddington: The Last Renaissance Biologist? Nat. Rev. Genet. 2002, 3, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Bak, P.; Tang, C.; Wiesenfeld, K. Self-Organized Criticality: An Explanation of the 1/f Noise. Phys. Rev. Lett. 1987, 59, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Keul, N.D.; Oruganty, K.; Schaper Bergman, E.T.; Beattie, N.R.; McDonald, W.E.; Kadirvelraj, R.; Gross, M.L.; Phillips, R.S.; Harvey, S.C.; Wood, Z.A. The Entropic Force Generated by Intrinsically Disordered Segments Tunes Protein Function. Nature 2018, 563, 584–588. [Google Scholar] [CrossRef]
- Erenpreisa, J. “Tamed” Chaos in Embryonal Development and Carcinogenesis: A Holistic View. Proc. Latv. Acad. Sci. Sect. B Nat. Exact Appl. Sci. 2000, 54, 1–8. [Google Scholar]
- Erenpreisa, J.; Zhukotsky, A. The Genome Field. Proc. Latv. Acad. Sci. Ser. B. 1992, 536, 64–68. [Google Scholar]
- Erenpreisa, J. Large Rosettes—The Element of the Suprachromonemal Organisation of Interphase Cell Nucleus. Proc. Latv. Acad. Sci. Ser. B. 1989, 7, 68–71. [Google Scholar]
- Erenpreisa, J. Two Mechanisms of Chromatin Compaction. Acta Histochem. 1989, 86, 129–135. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Zhukotsky, A. Interphase Genome as the Active Space: Chromatin Dynamics during Chick Embryo Chondrogenesis. Mech. Ageing Dev. 1993, 67, 21–32. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Zhukotsky, A.; Kozlov, A. The Chromatin Network: Image Analysis of Differentiating Chick Embryo Chondrocytes. Eur. J. Histochem. 1993, 37, 139–147. [Google Scholar]
- Schwarz-Finsterle, J.; Scherthan, H.; Huna, A.; González, P.; Mueller, P.; Schmitt, E.; Erenpreisa, J.; Hausmann, M. Volume Increase and Spatial Shifts of Chromosome Territories in Nuclei of Radiation-Induced Polyploidizing Tumour Cells. Mutat. Res. 2013, 756, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K. Field Hypothesis on the Self-Regulation of Gene Expression. J. Biol. Phys. 2002, 28, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Prigogine, I.; Stengers, I. Order Out of Chaos: Man’s New Dialogue with Nature; Bantam Books: New York, NY, USA, 1984; ISBN 9780553340822. [Google Scholar]
- Noble, D. Dance to the Tune of Life: Biological Relativity; Cambridge University Press: Cambridge, UK, 2016; ISBN 978-1107176249. [Google Scholar]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin Drives Compartmentalization of Inverted and Conventional Nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “Virgin Birth”, Polyploidy, and the Origin of Cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, C. Chromosome Territories, Nuclear Architecture and Gene Regulation in Mammalian Cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Abe, J.; Hiyama, T.B.; Mukaiyama, A.; Son, S.; Mori, T.; Saito, S.; Osako, M.; Wolanin, J.; Yamashita, E.; Kondo, T.; et al. Circadian Rhythms. Atomic-Scale Origins of Slowness in the Cyanobacterial Circadian Clock. Science 2015, 349, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.J.; Crew, R.C.; Wharfe, M.D.; Waddell, B.J. Rhythmic Three-Part Harmony: The Complex Interaction of Maternal, Placental and Fetal Circadian Systems. J. Biol. Rhythms 2017, 32, 534–549. [Google Scholar] [CrossRef]
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880. [Google Scholar] [CrossRef]
- Larsson, A.J.M.; Johnsson, P.; Hagemann-Jensen, M.; Hartmanis, L.; Faridani, O.R.; Reinius, B.; Segerstolpe, Å.; Rivera, C.M.; Ren, B.; Sandberg, R. Genomic Encoding of Transcriptional Burst Kinetics. Nature 2019, 565, 251–254. [Google Scholar] [CrossRef]
- Beckman, W.F.; Jiménez, M.Á.L.; Moerland, P.D.; Westerhoff, H.V.; Verschure, P.J. 4sUDRB-Sequencing for Genome-Wide Transcription Bursting Quantification in Breast Cancer Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Maass, P.G.; Barutcu, A.R.; Rinn, J.L. Interchromosomal Interactions: A Genomic Love Story of Kissing Chromosomes. J. Cell Biol. 2019, 218, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Krigerts, J.; Salmina, K.; Gerashchenko, B.I.; Freivalds, T.; Kurg, R.; Winter, R.; Krufczik, M.; Zayakin, P.; Hausmann, M.; et al. Heterochromatin Networks: Topology, Dynamics, and Function (a Working Hypothesis). Cells 2021, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Budylin, A. Related Changes in RNA Synthesis and DNA Superhelicity during Starvation of Ehrlich Ascites Tumour Cells. Proc. Latv. Acad. Sci. Sect. B Nat. Exact Appl. Sci. 1990, 10, 90–94. [Google Scholar]
- Budylin, A.; Erenpreisa, J.; Zhukotsky, A.; Zaleskaya, N. Fluctuations of the Reparable DNA Strand Breaks in Starving Tumour Cells. Proc. Latv. Acad. Sci. Sect. B Nat. Exact Appl. Sci. 1993, 5, 72–73. [Google Scholar]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return Journey from Cell Death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef]
- Saeki, Y.; Endo, T.; Ide, K.; Nagashima, T.; Yumoto, N.; Toyoda, T.; Suzuki, H.; Hayashizaki, Y.; Sakaki, Y.; Okada-Hatakeyama, M. Ligand-Specific Sequential Regulation of Transcription Factors for Differentiation of MCF-7 Cells. BMC Genom. 2009, 10, 545. [Google Scholar] [CrossRef] [Green Version]
- Krigerts, J.; Salmina, K.; Freivalds, T.; Zayakin, P.; Rumnieks, F.; Inashkina, I.; Giuliani, A.; Hausmann, M.; Erenpreisa, J. Differentiating Cancer Cells Reveal Early Large-Scale Genome Regulation by Pericentric Domains. Biophys. J. 2021, 120, 711–724. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Giuliani, A.; Zimatore, G.; Erenpreisa, J.; Yoshikawa, K. A Unified Genomic Mechanism of Cell-Fate Change. bioRxiv 2021. [Google Scholar] [CrossRef]
- Langton, C.G. Computation at the Edge of Chaos: Phase Transitions and Emergent Computation. Phys. D 1990, 42, 12–37. [Google Scholar] [CrossRef] [Green Version]
- Farshadi, E.; van der Horst, G.T.J.; Chaves, I. Molecular Links between the Circadian Clock and the Cell Cycle. J. Mol. Biol. 2020, 432, 3515–3524. [Google Scholar] [CrossRef] [PubMed]
- Neganova, I.; Lako, M. G1 to S Phase Cell Cycle Transition in Somatic and Embryonic Stem Cells. J. Anat. 2008, 213, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.-H.; Zhao, T.; Cui, K.; Hu, G.; Chen, Q.; Chen, W.; Wang, X.-W.; Soto-Gutierrez, A.; Zhao, K.; Deng, C.-X. Negative Reciprocal Regulation between Sirt1 and Per2 Modulates the Circadian Clock and Aging. Sci. Rep. 2016, 6, 28633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 Contributes to Telomere Maintenance and Augments Global Homologous Recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, P.-E.; Guilly, M.-N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor Cells Can Escape DNA-Damaging Cisplatin through DNA Endoreduplication and Reversible Polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Kosmacek, E.A.; Cragg, M.S.; Ianzini, F.; Anisimov, A.P. Polyploid Tumour Cells Elicit Paradiploid Progeny through Depolyploidizing Divisions and Regulated Autophagic Degradation. Cell Biol. Int. 2011, 35, 687–695. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-Induced Reprogramming of Breast Cancer Cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-Regulation of the Embryonic Self-Renewal Network through Reversible Polyploidy in Irradiated p53-Mutant Tumour Cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [Green Version]
- Shafi, A.A.; Knudsen, K.E. Cancer and the Circadian Clock. Cancer Res. 2019, 79, 3806–3814. [Google Scholar] [CrossRef] [Green Version]
- Shilts, J.; Chen, G.; Hughey, J.J. Evidence for Widespread Dysregulation of Circadian Clock Progression in Human Cancer. PeerJ 2018, 6, e4327. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Kettner, N.M. The Circadian Clock in Cancer Development and Therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar] [PubMed] [Green Version]
- Rijo-Ferreira, F.; Takahashi, J.S. Genomics of Circadian Rhythms in Health and Disease. Genome Med. 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer Res. 2019, 5, 475–494. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute Quantification of Somatic DNA Alterations in Human Cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef]
- Ilık, İ.A.; Aktaş, T. Nuclear Speckles: Dynamic Hubs of Gene Expression Regulation. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Kurg, R.; Krigerts, J.; Erenpreisa, J. The Topological Relationship between Ribogenesis, mRNA Transcription/splicing and the Tension of Actin Cytoskeleton. Biopolym. Cell 2019, 35, 231–232. [Google Scholar] [CrossRef]
- Philimonenko, V.V.; Zhao, J.; Iben, S.; Dingová, H.; Kyselá, K.; Kahle, M.; Zentgraf, H.; Hofmann, W.A.; de Lanerolle, P.; Hozák, P.; et al. Nuclear Actin and Myosin I Are Required for RNA Polymerase I Transcription. Nat. Cell Biol. 2004, 6, 1165–1172. [Google Scholar] [CrossRef]
- Wei, M.; Fan, X.; Ding, M.; Li, R.; Shao, S.; Hou, Y.; Meng, S.; Tang, F.; Li, C.; Sun, Y. Nuclear Actin Regulates Inducible Transcription by Enhancing RNA Polymerase II Clustering. Sci Adv 2020, 6, eaay6515. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Wu, S.; Hernandez, N. A Role for Beta-Actin in RNA Polymerase III Transcription. Genes Dev. 2004, 18, 3010–3015. [Google Scholar] [CrossRef] [Green Version]
- Burman, B.; Misteli, T.; Pegoraro, G. Quantitative Detection of Rare Interphase Chromosome Breaks and Translocations by High-Throughput Imaging. Genome Biol. 2015, 16, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozubek, S.; Lukásová, E.; Marecková, A.; Skalníková, M.; Kozubek, M.; Bártová, E.; Kroha, V.; Krahulcová, E.; Slotová, J. The Topological Organization of Chromosomes 9 and 22 in Cell Nuclei Has a Determinative Role in the Induction of t(9,22) Translocations and in the Pathogenesis of t(9,22) Leukemias. Chromosoma 1999, 108, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Lukášová, E.; Kozubek, S.; Kozubek, M.; Kjeronská, J.; Rýznar, L.; Horáková, J.; Krahulcová, E.; Horneck, G. Localisation and Distance between ABL and BCR Genes in Interphase Nuclei of Bone Marrow Cells of Control Donors and Patients with Chronic Myeloid Leukaemia. Hum. Genet. 1997, 100, 525–535. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Stringer, J.R.; Blough, R.; Medvedovic, M.; Fagin, J.A.; Nikiforov, Y.E. Proximity of Chromosomal Loci That Participate in Radiation-Induced Rearrangements in Human Cells. Science 2000, 290, 138–141. [Google Scholar] [CrossRef]
- Neves, H.; Ramos, C.; Gomes Da Silva, M.; Parreira, A.; Parreira, L. The Nuclear Topography of ABL, BCR, PML, and RARα Genes: Evidence for Gene Proximity in Specific Phases of the Cell Cycle and Stages of Hematopoietic Differentiation. Blood 1999, 93, 1197–1207. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Kozubek, S. Higher-Order Chromatin Structure in DSB Induction, Repair and Misrepair. Mutat. Res. Mutat. Res. 2010, 704, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M.; Lukasova, E.; Biswas, A.; Hildenbrand, G.; Davidkova, M.; Krasavin, E.; Kleibl, Z.; Falkova, I.; Jezkova, L.; et al. Determining Omics Spatiotemporal Dimensions Using Exciting New Nanoscopy Techniques to Assess Complex Cell Responses to DNA Damage: PART A-Radiomics. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M.; Lukasova, E.; Biswas, A.; Hildenbrand, G.; Davidkova, M.; Krasavin, E.; Kleibl, Z.; Falkova, I.; Jezkova, L.; et al. Determining Omics Spatiotemporal Dimensions Using Exciting New Nanoscopy Techniques to Assess Complex Cell Responses to DNA Damage: Part—Structuromics. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 225–247. [Google Scholar] [CrossRef]
- Falk, M.; Hausmann, M. A Paradigm Revolution or Just Better Resolution—Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation? Cancers 2021, 13, 18. [Google Scholar] [CrossRef]
- Kozubek, S.; Lukásová, E.; Jirsová, P.; Koutná, I.; Kozubek, M.; Ganová, A.; Bártová, E.; Falk, M.; Paseková, R. 3D Structure of the Human Genome: Order in Randomness. Chromosoma 2002, 111, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukásová, E.; Kozubek, S.; Kozubek, M. Topography of Genetic Elements of X-Chromosome Relative to the Cell Nucleus and to the Chromosome X Territory Determined for Human Lymphocytes. Gene 2002, 292, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Lukásová, E.; Kozubek, S.; Kozubek, M.; Falk, M.; Amrichová, J. The 3D Structure of Human Chromosomes in Cell Nuclei. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2002, 10, 535–548. [Google Scholar] [CrossRef]
- Misteli, T. Higher-Order Genome Organization in Human Disease. Cold Spring Harb. Perspect. Biol. 2010, 2, a000794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancaldi, V. Chromatin Network Analyses: Towards Structure-Function Relationships in Epigenomics. Front. Bioinform. 2021, 1, 742216. [Google Scholar] [CrossRef]
- Raynaud, C.; Mallory, A.C.; Latrasse, D.; Jégu, T.; Bruggeman, Q.; Delarue, M.; Bergounioux, C.; Benhamed, M. Chromatin Meets the Cell Cycle. J. Exp. Bot. 2014, 65, 2677–2689. [Google Scholar] [CrossRef] [Green Version]
- Chapski, D.J.; Rosa-Garrido, M.; Hua, N.; Alber, F.; Vondriska, T.M. Spatial Principles of Chromatin Architecture Associated with Organ-Specific Gene Regulation. Front. Cardiovasc. Med. 2019, 5, 186. [Google Scholar] [CrossRef]
- Winick-Ng, W.; Kukalev, A.; Harabula, I.; Zea-Redondo, L.; Szabó, D.; Meijer, M.; Serebreni, L.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. Cell-Type Specialization Is Encoded by Specific Chromatin Topologies. Nature 2021, 599, 684–691. [Google Scholar] [CrossRef]
- Zhang, K.; Hocker, J.D.; Miller, M.; Hou, X.; Chiou, J.; Poirion, O.B.; Qiu, Y.; Li, Y.E.; Gaulton, K.J.; Wang, A.; et al. A Single-Cell Atlas of Chromatin Accessibility in the Human Genome. Cell 2021, 184, 5985–6001.e19. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Chromatin Dynamics during DSB Repair. Biochim. Biophys. Acta 2007, 1773, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirman, M.J.; Gill, H.J.; Burnett, R.C.; Mbangkollo, D.; McCabe, N.R.; Kobayashi, H.; Ziemin-van der Poel, S.; Kaneko, Y.; Morgan, R.; Sandberg, A.A.; et al. Rearrangement of the MLL Gene in Acute Lymphoblastic and Acute Myeloid Leukemias with 11q23 Chromosomal Translocations. N. Engl. J. Med. 1993, 329, 909–914. [Google Scholar] [CrossRef]
- Jia, Y.; Li, C.; Zhao, J.; Song, Y.; Wang, J.; Mi, Y. The Discussion of t(1;17)(P11;Q21) Translocation in Acute Promyelocytic Leukemia Patient on Molecular Remission. Clin. Case Rep. 2017, 5, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Pagáčová, E.; Falk, M.; Falková, I.; Lukášová, E.; Michalová, K.; Oltová, A.; Raška, I.; Kozubek, S. Frequent Chromatin Rearrangements in Myelodysplastic Syndromes—What Stands Behind? Folia Biol. (Praha) 2014, 60 (Suppl. S1), 1–7. [Google Scholar] [PubMed]
- Berendes, P.; Hoogeveen, A.; van Dijk, M.; van Denderen, J.; van Ewijk, W. Specific immunologic recognition of the tumor-specific E2A-PBX1 fusion-point antigen in t(1;19)-positive pre-B cells. Leukemia 1995, 9, 1321–1327. [Google Scholar]
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with Similar LET Values Generate DNA Breaks of Different Complexity and Reparability: A High-Resolution Microscopy Analysis of Γh2AX/53BP1 Foci. Nanoscale 2018, 10, 1162–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Lin, L.F.; Hahn, J.; Xie, J.; Holman, H.F.; Yuan, C. Single-Cell Image-Based Analysis Reveals Chromatin Changes during the Acquisition of Tamoxifen Drug Resistance. Life 2022, 12, 438. [Google Scholar] [CrossRef]
- Falk, M.; Lukášová, E.; Štefančíková, L.; Baranová, E.; Falková, I.; Ježková, L.; Davídková, M.; Bačíková, A.; Vachelová, J.; Michaelidesová, A.; et al. Heterochromatinization Associated with Cell Differentiation as a Model to Study DNA Double Strand Break Induction and Repair in the Context of Higher-Order Chromatin Structure. Appl. Radiat. Isot. 2014, 83, 177–185. [Google Scholar] [CrossRef]
- Falk, M.; Lukasova, E.; Falkova, I.; Stefancikova, L.; Jezkova, L.; Bacikova, A.; Davidkova, M.; Boryeko, A.; Krasavin, E.A.; Kozubek, S. Chromatin Differentiation of White Blood Cells Decreases DSB Damage Induction, Prevents Functional Assembly of Repair Foci, but Has No Influence on Protrusion of Heterochromatic DSBs into the Low-Dense Chromatin. J. Rad. Res. 2014, 55, i81–i82. [Google Scholar] [CrossRef]
- Kozubek, S.; Lukásová, E.; Rýznar, L.; Kozubek, M.; Lisková, A.; Govorun, R.D.; Krasavin, E.A.; Horneck, G. Distribution of ABL and BCR Genes in Cell Nuclei of Normal and Irradiated Lymphocytes. Blood 1997, 89, 4537–4545. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yu, R.; Dang, W. Chromatin Architectural Changes during Cellular Senescence and Aging. Genes 2018, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Feser, J.; Tyler, J. Chromatin Structure as a Mediator of Aging. FEBS Lett. 2011, 585, 2041–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukasova, E.; Koristek, Z.; Falk, M.; Kozubek, S.; Grigoryev, S.; Kozubek, M.; Ondrej, V.; Kroupová, I. Methylation of Histones in Myeloid Leukemias as a Potential Marker of Granulocyte Abnormalities. J. Leukoc. Biol. 2005, 77, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Lukasova, E.; Kozubek, S.; Falk, M.; Kozubek, M.; Zaloudík, J.; Vagunda, V.; Pavlovský, Z. Topography of Genetic Loci in the Nuclei of Cells of Colorectal Carcinoma and Adjacent Tissue of Colonic Epithelium. Chromosoma 2004, 112, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Bártová, E.; Kozubek, S.; Kozubek, M.; Jirsová, P.; Lukášová, E.; Skalníková, M.; Buchníčková, K. The Influence of the Cell Cycle, Differentiation and Irradiation on the Nuclear Location of the Abl, Bcr and c-Myc Genes in Human Leukemic Cells. Leuk. Res. 2000, 24, 233–241. [Google Scholar] [CrossRef]
- Ismail, S.I.; Naffa, R.G.; Yousef, A.-M.F.; Ghanim, M.T. Incidence of Bcr-Abl Fusion Transcripts in Healthy Individuals. Mol. Med. Rep. 2014, 9, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Quina, A.S.; Gameiro, P.; Sá da Costa, M.; Telhada, M.; Parreira, L. PML-RARA Fusion Transcripts in Irradiated and Normal Hematopoietic Cells. Genes Chromosom. Cancer 2000, 29, 266–275. [Google Scholar] [CrossRef]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in Chromatin Structure and Mobility in Living Cells at Sites of DNA Double-Strand Breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Aten, J.A.; Stap, J.; Krawczyk, P.M.; van Oven, C.H.; Hoebe, R.A.; Essers, J.; Kanaar, R. Dynamics of DNA Double-Strand Breaks Revealed by Clustering of Damaged Chromosome Domains. Science 2004, 303, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Cremer, T.; Cremer, M. Chromosome Territories. Cold Spring Harb. Perspect. Biol. 2010, 2, a003889. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Su, J.-H.; Beliveau, B.J.; Bintu, B.; Moffitt, J.R.; Wu, C.; Zhuang, X. Spatial Organization of Chromatin Domains and Compartments in Single Chromosomes. Science 2016, 353, 598–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tena, J.J.; Santos-Pereira, J.M. Topologically Associating Domains and Regulatory Landscapes in Development, Evolution and Disease. Front. Cell Dev. Biol. 2021, 9, 702787. [Google Scholar] [CrossRef] [PubMed]
- Gümüş, G.; Sunguroğlu, A.; Tükün, A.; Sayin, D.B.; Bökesoy, I. Common Fragile Sites Associated with the Breakpoints of Chromosomal Aberrations in Hematologic Neoplasms. Cancer Genet. Cytogenet. 2002, 133, 168–171. [Google Scholar] [CrossRef]
- de Lima, M.F.; Lisboa, M.d.O.; Terceiro, L.E.L.; Rangel-Pozzo, A.; Mai, S. Chromosome Territories in Hematological Malignancies. Cells 2022, 11, 1368. [Google Scholar] [CrossRef]
- Kozubek, S.; Lukášová, E.; Rýznar, L.; Kozubek, M.; Lišková, A.; Kroha, V. Chromatin Structure and Its Cell Cycle Kinetics in Intact and Irradiated Cell Nuclei. In Fundamentals for the Assessment of Risks from Environmental Radiation; Baumstark-Khan, C., Kozubek, S., Horneck, G., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 231–242. ISBN 978-0-7923-5668-4. [Google Scholar]
- Mani, R.-S.; Tomlins, S.A.; Callahan, K.; Ghosh, A.; Nyati, M.K.; Varambally, S.; Palanisamy, N.; Chinnaiyan, A.M. Induced Chromosomal Proximity and Gene Fusions in Prostate Cancer. Science 2009, 326, 1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Z.; Mai, H.; Liu, X.; Fu, X.; Zhang, X.; Xie, L.; Chen, Y.; Shlien, A.; Wen, F. Single-Cell Diploid Hi-C Reveals the Role of Spatial Aggregations in Complex Rearrangements and KMT2A Fusions in Leukemia. Genome Biol. 2022, 23, 173. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-Dimensional Genome Architecture Influences Partner Selection for Chromosomal Translocations in Human Disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef]
- Zadneprianetc, M.; Boreyko, A.; Jezkova, L.; Falk, M.; Ryabchenko, A.; Hramco, T.; Krupnova, M.; Kulikova, E.; Pavlova, A.; Shamina, D.; et al. Clustered DNA Damage Formation in Human Cells after Exposure to Low- and Intermediate-Energy Accelerated Heavy Ions. Phys. Part. Nucl. Lett. 2022, 19, 440–450. [Google Scholar] [CrossRef]
- Gyurkó, D.M.; Veres, D.V.; Módos, D.; Lenti, K.; Korcsmáros, T.; Csermely, P. Adaptation and Learning of Molecular Networks as a Description of Cancer Development at the Systems-Level: Potential Use in Anti-Cancer Therapies. Semin. Cancer Biol. 2013, 23, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Ježková, L.; Falk, M.; Falková, I.; Davídková, M.; Bačíková, A.; Štefančíková, L.; Vachelová, J.; Michaelidesová, A.; Lukášová, E.; Boreyko, A.; et al. Function of Chromatin Structure and Dynamics in DNA Damage, Repair and Misrepair: γ-Rays and Protons in Action. Appl. Radiat. Isot. 2014, 83, 128–136. [Google Scholar] [CrossRef]
- Walen, K.H. Normal human cell conversion to 3-D cancer-like growth: Genome damage, endopolyploidy, senecence escape, and cell polarity change/loss. J. Cancer Ther. 2011, 2, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin Structure Influences the Sensitivity of DNA to Gamma-Radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Local Changes of Higher-Order Chromatin Structure during DSB-Repair. J. Phys. Conf. Ser. 2008, 101, 012018. [Google Scholar] [CrossRef]
- Koeffler, H.P.; Golde, D.W. Chronic Myelogenous Leukemia—New Concepts. N. Engl. J. Med. 1981, 304, 1201–1209. [Google Scholar] [CrossRef]
- Kang, Z.-J.; Liu, Y.-F.; Xu, L.-Z.; Long, Z.-J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.-X.; Pan, Y.-J.; Yan, J.-S.; et al. The Philadelphia Chromosome in Leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [Green Version]
- Massimino, M.; Tirrò, E.; Stella, S.; Manzella, L.; Pennisi, M.S.; Romano, C.; Vitale, S.R.; Puma, A.; Tomarchio, C.; Di Gregorio, S.; et al. Impact of the Breakpoint Region on the Leukemogenic Potential and the TKI Responsiveness of Atypical BCR-ABL1 Transcripts. Front. Pharmacol. 2021, 12, 669469. [Google Scholar] [CrossRef] [PubMed]
- Wilde, L.; Cooper, J.; Wang, Z.-X.; Liu, J. Clinical, Cytogenetic, and Molecular Findings in Two Cases of Variant t(8;21) Acute Myeloid Leukemia (AML). Front. Oncol. 2019, 9, 1016. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.V. The Molecular Biology of Chronic Myeloid Leukaemia. Leukemia 1996, 10, 751–756. [Google Scholar]
- Pourrajab, F.; Zare-Khormizi, M.R.; Hashemi, A.S.; Hekmatimoghaddam, S. Genetic Characterization and Risk Stratification of Acute Myeloid Leukemia. Cancer Manag. Res. 2020, 12, 2231–2253. [Google Scholar] [CrossRef] [Green Version]
- Rosin, L.F.; Crocker, O.; Isenhart, R.L.; Nguyen, S.C.; Xu, Z.; Joyce, E.F. Chromosome Territory Formation Attenuates the Translocation Potential of Cells. eLife 2019, 8, e49553. [Google Scholar] [CrossRef]
- Cafourková, A.; Luká ová, E.; Kozubek, S.; Kozubek, M.; Govorun, R.D.; Koutná, I.; Bártová, E.; Skalníková, M.; Jirsová, P.; Paseková, R.; et al. Exchange Aberrations among 11 Chromosomes of Human Lymphocytes Induced by Gamma-Rays. Int. J. Radiat. Biol. 2001, 77, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Ebert, B.L. The Biology and Clinical Impact of Genetic Lesions in Myeloid Malignancies. Blood 2013, 122, 3741–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abáigar, M.; Robledo, C.; Benito, R.; Ramos, F.; Díez-Campelo, M.; Hermosín, L.; Sánchez-del-Real, J.; Alonso, J.M.; Cuello, R.; Megido, M.; et al. Chromothripsis Is a Recurrent Genomic Abnormality in High-Risk Myelodysplastic Syndromes. PLoS ONE 2016, 11, e0164370. [Google Scholar] [CrossRef] [PubMed]
- Lukášová, E.; Kořistek, Z.; Klabusay, M.; Ondřej, V.; Grigoryev, S.; Bačíková, A.; Řezáčová, M.; Falk, M.; Vávrová, J.; Kohútová, V.; et al. Granulocyte Maturation Determines Ability to Release Chromatin NETs and Loss of DNA Damage Response; These Properties Are Absent in Immature AML Granulocytes. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 767–779. [Google Scholar] [CrossRef] [Green Version]
- Depes, D.; Lee, J.-H.; Bobkova, E.; Jezkova, L.; Falkova, I.; Bestvater, F.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; et al. Single-Molecule Localization Microscopy as a Promising Tool for γH2AX/53BP1 Foci Exploration. Eur. Phys. J. D 2018, 72, 158. [Google Scholar] [CrossRef]
- Bobkova, E.; Depes, D.; Lee, J.-H.; Jezkova, L.; Falkova, I.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; Kulikova, E.; et al. Recruitment of 53BP1 Proteins for DNA Repair and Persistence of Repair Clusters Differ for Cell Types as Detected by Single Molecule Localization Microscopy. Int. J. Mol. Sci. 2018, 19, 3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Hausmann, M. Advances in Research of DNA Damage and Repair in Cells Exposed to Various Types of Ionizing Radiation in the Era of Super-Resolution Optical Microscopy. Cas. Lek. Cesk. 2020, 159, 286–297. [Google Scholar]
- Hausmann, M.; Neitzel, C.; Bobkova, E.; Nagel, D.; Hofmann, A.; Chramko, T.; Smirnova, E.; Kopečná, O.; Pagáčová, E.; Boreyko, A.; et al. Single Molecule Localization Microscopy Analyses of DNA-Repair Foci and Clusters Detected Along Particle Damage Tracks. Front. Phys. 2020, 8, 578662. [Google Scholar] [CrossRef]
- Hahn, H.; Neitzel, C.; Kopečná, O.; Heermann, D.W.; Falk, M.; Hausmann, M. Topological Analysis of γH2AXand MRE11 Clusters Detected by Localization Microscopy during X-ray Induced DNA Double-Strand Break Repair. Cancers 2021, 13, 5561. [Google Scholar] [CrossRef]
- Vicar, T.; Gumulec, J.; Kolar, R.; Falkova, I.; Kopecna, O.; Pagacova, E.; Falk, M. Advanced Learning- Based Algorithm for Fast Automatic Analysis of DNA Double Strand Break (DSB) Repair Foci. Comput. Struct. Biotechnol. J. 2021, 19, 6465–6480. [Google Scholar] [CrossRef]
- Schroedinger, E. What Is Life? The Physical Aspect of the Living Cell, Etc; Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Bizzarri, M.; Naimark, O.; Nieto-Villar, J.; Fedeli, V.; Giuliani, A. Complexity in biological organization: Deconstruction (and subsequent restating) of key concepts. Entropy 2020, 22, 885. [Google Scholar] [CrossRef]
- Cherstvy, A.G.; Teif, V.B. Structure-driven homology pairing of chromatin fibers: The role of electrostatics and protein-induced bridging. J. Biol. Phys. 2013, 39, 363–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krufczik, M.; Sievers, A.; Hausmann, A.; Lee, J.-H.; Hildenbrand, G.; Schaufler, W.; Hausmann, M. Combining low temperature fluorescence DNA-hybridization, immunostaining, and super-resolution localization microscopy for nano-structure analysis of ALU elements and their influence on chromatin structure. Int. J. Mol. Sci. 2017, 18, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Laure Djikimi Tchetgna, F.; Krufczik, M.; Schmitt, E.; Cremer, C.; Bestvater, F.; Hausmann, M. COMBO-FISH: A versatile tool beyond standard FISH to study chromatin organization by fluorescence light microscopy. OBM Genet. 2019, 3, 1. [Google Scholar] [CrossRef]
- Gorban, A.N.; Tyukina, T.A.; Pokidysheva, L.I.; Smirnova, E.V. Dynamic and thermodynamic models of adaptation. Phys. Life Rev. 2021, 37, 17–64. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Hausmann, M. Super-Resolution Radiation Biology: From Bio-Dosimetry towards Nano-Studies of DNA Repair Mechanisms. In DNA-Repair; Behzadi, P., Ed.; Intech-Open: Rijeka, Croatia, 2020; ISBN 978-1-83881-094-8. [Google Scholar] [CrossRef]
- Hausmann, M.; Neitzel, C.; Hahn, H.; Winter, R.; Falkova, I.; Heermann, D.W.; Pilarczyk, G.; Hildenbrand, G.; Scherthan, H.; Falk, M. Space and time in the universe of the cell nucleus after ionizing radiation attacks: A comparison of cancer and non-cancer cell response. (presented on 1st Int. Elec. Conf. Cancers: Exploiting Cancer Vulnerability by Targeting the DNA Damage Response). Med. Sci. Forum 2021, 3, 15. [Google Scholar]
- Ripley, B.D. Modelling Spatial Patterns. J. R. Stat. Soc. Ser. B Methodol. 1977, 39, 172–192. [Google Scholar] [CrossRef]
- Hofmann, A.; Krufczik, M.; Heermann, D.W.; Hausmann, M. Using Persistent Homology as a New Approach for Super-Resolution Localization Microscopy Data Analysis and Classification of γ2AX Foci/Clusters. Int. J. Mol. Sci. 2018, 19, 2263. [Google Scholar]
- Morales, M.E.; White, T.B.; Streva, V.A.; DeFreece, C.B.; Hedges, D.J.; Deininger, P.L. The contribution of ALU elements to mutagenic DNA double-strand break repair. PLoS Genet. 2015, 11, e1005016. [Google Scholar] [CrossRef]
- Gu, Z.; Jin, K.; Crabbe, M.J.C.; Zhang, Y.; Liu, X.; Huang, Y.; Hua, M.; Nan, P.; Zhang, Z.; Zhong, Y. Enrichment analysis of ALU elements with different spatial chromatin proximity in the human genome. Protein Cell 2016, 7, 250–266. [Google Scholar] [CrossRef]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation induced chromatin conformation changes analysed by fluorescent localization microscopy, statistical physics, and graph theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Heermann, D.W. DNA double-strand breaks: Linking gene expression to chromosome morphology and mobility. Chromosoma 2014, 123, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, M.; Ilíc, N.; Pilarczyk, G.; Lee, J.-H.; Logeswaran, A.; Borroni, A.P.; Krufczik, M.; Theda, F.; Waltrich, N.; Bestvater, F.; et al. Challenges for super-resolution localization microscopy and biomolecular fluorescent nano-probing in cancer research. Int. J. Mol. Sci. 2017, 18, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausmann, M.; Wagner, E.; Lee, J.-H.; Schrock, G.; Schaufler, W.; Krufczik, M.; Papenfuß, F.; Port, M.; Bestvater, F.; Scherthan, H. Super-resolution localization microscopy of radiation-induced histone H2AX-phosphorylation in relation to H3K9-trimethylation in HeLa cells. Nanoscale 2018, 10, 4320–4331. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, M.; Falk, M.; Neitzel, C.; Hofmann, A.; Biswas, A.; Gier, T.; Falkova, I.; Heermann, D.W.; Hildenbrand, G. Elucidation of the clustered nano-architecture of radiation-induced DNA damage sites and surrounding chromatin in cancer cells: A Single Molecule Localization Microscopy approach. Int. J. Mol. Sci. 2021, 22, 3636. [Google Scholar] [CrossRef] [PubMed]
- Scherthan, H.; Lee, J.-H.; Maus, E.; Schumann, S.; Muhtadi, R.; Chojowski, R.; Port, M.; Lassmann, M.; Bestvater, F.; Hausmann, M. Nanostructure of Clustered DNA Damage in Leukocytes after In-Solution Irradiation with the Alpha Emitter Ra. Cancers 2019, 11, 1877. [Google Scholar] [CrossRef] [Green Version]
- Livolant, F. Ordered Phases of DNA in vivo and in vitro. Phys. A Stat. Mech. Its Appl. 1991, 176, 117–137. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Murphy, L.D. Macromolecular Crowding and the Mandatory Condensation of DNA in Bacteria. FEBS Lett. 1996, 390, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Bloomfield, V.A. DNA Condensation. Curr. Opin. Struct. Biol. 1996, 6, 334–341. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Yoshikawa, Y. Compaction and Condensation of DNA. In Pharmaceutical Perspectives of Nucleic Acid-Based Therapy; Mahato, R.I., Kim, S.W., Eds.; Taylor & Francis: London, UK, 2002; Chapter 8; pp. 136–163. ISBN 0-415-28385-X. [Google Scholar]
- Schiessel, H. The Physics of Chromatin. J. Phys. Condens. Matter 2003, 15, R699. [Google Scholar] [CrossRef]
- Zinchenko, A.A.; Pyshkina, O.A.; Andrey, L.V.; Sergeyev, V.G.; Yoshikawa, K. Single DNA molecules: Compaction and decompaction. In DNA Interactions with Polymers and Surfactants; Dias, R., Lindman, B., Eds.; Wiley: Hoboken, NJ, USA, 2008; Chapter 3; p. 59. ISBN 978-0-470-28635-7. [Google Scholar]
- de Gennes, P.-G.; Gennes, P.-G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979; ISBN 9780801412035. [Google Scholar]
- Khokhlov, A.R.; Grosberg, A.Y.; Paride, V.S. Statistical Physics of Macromolecules; AIP Press: New York, NY, USA, 1994; ISBN 9781563960710. [Google Scholar]
- Estévez-Torres, A.; Baigl, D. DNA Compaction: Fundamentals and Applications. Soft Matter 2011, 7, 6746–6756. [Google Scholar] [CrossRef]
- Minagawa, K.; Matsuzawa, Y.; Yoshikawa, K.; Khokhlov, A.R.; Doi, M. Direct Observation of the Coil-Globule Transition in Dna Molecules. Biopolymers 1994, 34, 555–558. [Google Scholar] [CrossRef]
- Mel’nikov, S.M.; Sergeyev, V.G.; Yoshikawa, K. Discrete Coil-Globule Transition of Large DNA Induced by Cationic Surfactant. J. Am. Chem. Soc. 1995, 117, 2401–2408. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Takahashi, M.; Vasilevskaya, V.V.; Khokhlov, A.R. Large Discrete Transition in a Single DNA Molecule Appears Continuous in the Ensemble. Phys. Rev. Lett. 1996, 76, 3029–3031. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yoshikawa, K.; Vasilevskaya, V.V.; Khokhlov, A.R. Discrete Coil−Globule Transition of Single Duplex DNAs Induced by Polyamines. J. Phys. Chem. B 1997, 101, 9396–9401. [Google Scholar] [CrossRef]
- Vasilevskaya, V.V.; Khokhlov, A.R.; Matsuzawa, Y.; Yoshikawa, K. Collapse of Single DNA Molecule in Poly(ethylene Glycol) Solutions. J. Chem. Phys. 1995, 102, 6595–6602. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Kidoaki, S.; Takahashi, M.; Vasilevskaya, V.V.; Khokhlov, A.R. Marked Discreteness on the Coil-Globule Transition of Single Duplex DNA. Ber. Bunsenges. Phys. Chem. 1996, 100, 876–880. [Google Scholar] [CrossRef]
- Tongu, C.; Kenmotsu, T.; Yoshikawa, Y.; Zinchenko, A.; Chen, N.; Yoshikawa, K. Divalent Cation Shrinks DNA but Inhibits Its Compaction with Trivalent Cation. J. Chem. Phys. 2016, 144, 205101. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.; Thomas, T.J. Polyamines in Cell Growth and Cell Death: Molecular Mechanisms and Therapeutic Applications. Cell. Mol. Life Sci. 2001, 58, 244–258. [Google Scholar] [CrossRef]
- Childs, A.C.; Mehta, D.J.; Gerner, E.W. Polyamine-Dependent Gene Expression. Cell. Mol. Life Sci. 2003, 60, 1394–1406. [Google Scholar] [CrossRef]
- Igarashi, K.; Kashiwagi, K. Modulation of Cellular Function by Polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef]
- Kanemura, A.; Yoshikawa, Y.; Fukuda, W.; Tsumoto, K.; Kenmotsu, T.; Yoshikawa, K. Opposite Effect of Polyamines on in vitro Gene Expression: Enhancement at Low Concentrations but Inhibition at High Concentrations. PLoS ONE 2018, 13, e0193595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, T.; Yoshikawa, Y.; Fukuda, W.; Umezawa, N.; Higuchi, T.; Fujiwara, S.; Imanaka, T.; Yoshikawa, K. Branched-Chain Polyamine Found in Hyperthermophiles Induces Unique Temperature-Dependent Structural Changes in Genome-Size DNA. Chemphyschem 2018, 19, 2299–2304. [Google Scholar] [CrossRef] [Green Version]
- Nishio, T.; Yoshikawa, Y.; Shew, C.-Y.; Umezawa, N.; Higuchi, T.; Yoshikawa, K. Specific Effects of Antitumor Active Norspermidine on the Structure and Function of DNA. Sci. Rep. 2019, 9, 14971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, T.; Nishio, T.; Yoshikawa, Y.; Umezawa, N.; Higuchi, T.; Shew, C.-Y.; Kenmotsu, T.; Yoshikawa, K. Effects of Structural Isomers of Spermine on the Higher-Order Structure of DNA and Gene Expression. Int. J. Mol. Sci. 2021, 22, 2355. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Yoshikawa, Y.; Yoshikawa, K.; Sato, S.-I. Longer DNA Exhibits Greater Potential for Cell-Free Gene Expression. Sci. Rep. 2021, 11, 11739. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Yoshikawa, K. Phase Transition and Phase Segregation in a Single Double-Stranded DNA Molecule. Phys. Rev. Lett. 1996, 77, 2133–2136. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Yoshikawa, Y.; Koyama, Y.; Kanbe, T. Highly Effective Compaction of Long Duplex DNA Induced by Polyethylene Glycol with Pendant Amino Groups. J. Am. Chem. Soc. 1997, 119, 6473–6477. [Google Scholar] [CrossRef]
- Takagi, S.; Tsumoto, K.; Yoshikawa, K. Intra-Molecular Phase Segregation in a Single Polyelectrolyte Chain. J. Chem. Phys. 2001, 114, 6942–6949. [Google Scholar] [CrossRef]
- Iwaki, T.; Yoshikawa, K. Competition between Interchain and Intrachain Phase Segregation. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003, 68, 031902. [Google Scholar] [CrossRef]
- Iwaki, T.; Yoshikawa, K. Counterion-Mediated Intra-Chain Phase Segregation on a Single Semiflexible Polyelectrolyte Chain. EPL 2004, 68, 113. [Google Scholar] [CrossRef]
- Nakai, T.; Hizume, K.; Yoshimura, S.H.; Takeyasu, K.; Yoshikawa, K. Phase Transition in Reconstituted Chromatin. EPL 2005, 69, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Hizume, K.; Araki, S.; Yoshikawa, K.; Takeyasu, K. Topoisomerase II, Scaffold Component, Promotes Chromatin Compaction in Vitro in a Linker-Histone H1-Dependent Manner. Nucleic Acids Res. 2007, 35, 2787–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hizume, K.; Nakai, T.; Araki, S.; Prieto, E.; Yoshikawa, K.; Takeyasu, K. Removal of Histone Tails from Nucleosome Dissects the Physical Mechanisms of Salt-Induced Aggregation, Linker Histone H1-Induced Compaction, and 30-Nm Fiber Formation of the Nucleosome Array. Ultramicroscopy 2009, 109, 868–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, T.; Sugino, K.; Yoshikawa, Y.; Matsumoto, M.; Oe, Y.; Sadakane, K.; Yoshikawa, K. K+ Promotes the Favorable Effect of Polyamine on Gene Expression Better than Na+. PLoS ONE 2020, 15, e0238447. [Google Scholar] [CrossRef]
- Tsumoto, K.; Yoshikawa, K. RNA Switches the Higher-Order Structure of DNA. Biophys. Chem. 1999, 82, 1–8. [Google Scholar] [CrossRef]
- Rudiuk, S.; Saito, H.; Hara, T.; Inoue, T.; Yoshikawa, K.; Baigl, D. Light-Regulated mRNA Condensation by a Photosensitive Surfactant Works as a Series Photoswitch of Translation Activity in the Presence of Small RNAs. Biomacromolecules 2011, 12, 3945–3951. [Google Scholar] [CrossRef]
- Akitaya, T.; Tsumoto, K.; Yamada, A.; Makita, N.; Kubo, K.; Yoshikawa, K. NTP Concentration Switches Transcriptional Activity by Changing the Large-Scale Structure of DNA. Biomacromolecules 2003, 4, 1121–1125. [Google Scholar] [CrossRef]
- Tsumoto, K.; Luckel, F.; Yoshikawa, K. Giant DNA Molecules Exhibit On/off Switching of Transcriptional Activity through Conformational Transition. Biophys. Chem. 2003, 106, 23–29. [Google Scholar] [CrossRef]
- Tsuji, A.; Yoshikawa, K. ON-OFF Switching of Transcriptional Activity of Large DNA through a Conformational Transition in Cooperation with Phospholipid Membrane. J. Am. Chem. Soc. 2010, 132, 12464–12471. [Google Scholar] [CrossRef]
- Tsumoto, K.; Sakuta, H.; Takiguchi, K.; Yoshikawa, K. Nonspecific Characteristics of Macromolecules Create Specific Effects in Living Cells. Biophys. Rev. 2020, 12, 425–434. [Google Scholar] [CrossRef]
- Takenaka, Y.; Hagahara, H.; Kitahata, H.; Yoshikawa, K. Large-scale on-off switching of genetic activity mediated by the folding-unfolding transition in a giant DNA molecule: An hypothesis. Phys. Rev. E 2008, 77, 31905. [Google Scholar] [CrossRef]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-Specific Polycomb Targets and de Novo DNA Methylation Define Restriction and Potential of Neuronal Progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, Silencing Potential and Evolutionary Impact of Promoter DNA Methylation in the Human Genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.-K.; Movassagh, M.; Goh, H.-G.; Bennett, M.R.; Down, T.A.; Foo, R.S.Y. Genome-Wide Conserved Consensus Transcription Factor Binding Motifs Are Hyper-Methylated. BMC Genom. 2010, 11, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlet, T.; Argüeso Lleida, A.; Al Adhami, H.; Dumas, M.; Bender, A.; Ngondo, R.P.; Tanguy, M.; Vallet, J.; Auclair, G.; Bardet, A.F.; et al. Genome-Wide Analysis in the Mouse Embryo Reveals the Importance of DNA Methylation for Transcription Integrity. Nat. Commun. 2020, 11, 3153. [Google Scholar] [CrossRef] [PubMed]
- Brivanlou, A.H.; Darnell, J.E., Jr. Signal Transduction and the Control of Gene Expression. Science 2002, 295, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Roeder, R.G. The Role of General Initiation Factors in Transcription by RNA Polymerase II. Trends Biochem. Sci. 1996, 21, 327–335. [Google Scholar] [CrossRef]
- Nikolov, D.B.; Burley, S.K. RNA Polymerase II Transcription Initiation: A Structural View. Proc. Natl. Acad. Sci. USA 1997, 94, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Davidkova, G.; Zhou, L.W. Antisense RNA Gene Therapy for Studying and Modulating Biological Processes. Cell. Mol. Life Sci. 1999, 55, 334–358. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, S.; Vidyarthi, A.S.; Prasad, D. RNA Interference: Concept to Reality in Crop Improvement. Planta 2014, 239, 543–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goñi, J.R.; Pérez, A.; Torrents, D.; Orozco, M. Determining Promoter Location Based on DNA Structure First-Principles Calculations. Genome Biol. 2007, 8, R263. [Google Scholar] [CrossRef] [Green Version]
- Meysman, P.; Marchal, K.; Engelen, K. DNA Structural Properties in the Classification of Genomic Transcription Regulation Elements. Bioinform. Biol. Insights 2012, 6, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Li, Y.; Shim, A.R.; Virk, R.K.A.; Agrawal, V.; Eshein, A.; Nap, R.J.; Almassalha, L.M.; Backman, V.; Szleifer, I. Physical and Data Structure of 3D Genome. Sci. Adv. 2020, 6, eaay4055. [Google Scholar] [CrossRef] [Green Version]
- Travers, A.; Muskhelishvili, G. DNA Structure and Function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef]
- Peters, J.P., 3rd; Maher, L.J. DNA Curvature and Flexibility in vitro and in vivo. Q. Rev. Biophys. 2010, 43, 23–63. [Google Scholar] [CrossRef]
- Sievers, A.; Sauer, L.; Hausmann, M.; Hildenbrand, G. Eukaryotic Genomes Show Strong Evolutionary Conservation of K-Mer Composition and Correlation Contributions between Introns and Intergenic Regions. Genes 2021, 12, 1571. [Google Scholar] [CrossRef]
- Sievers, A.; Bosiek, K.; Bisch, M.; Dreessen, C.; Riedel, J.; Froß, P.; Hausmann, M.; Hildenbrand, G. K-Mer Content, Correlation, and Position Analysis of Genome DNA Sequences for the Identification of Function and Evolutionary Features. Genes 2017, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Mojtahedi, M.; Skupin, A.; Zhou, J.; Castano, I.G.; Leong-Quong, R.Y.Y.; Chang, H.; Trachana, K.; Giuliani, A.; Huang, S. Cell fate decision as high-dimensional critical state transition. PLoS Biol. 2016, 14, e2000640. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Fu, X.; Ma, G.; Hutchins, A.P. Chromatin and epigenetic rearrangements in embryonic stem cell fate transitions. Front. Cell Dev. Biol. 2021, 9, 637309. [Google Scholar] [CrossRef] [PubMed]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erenpreisa, J.; Giuliani, A.; Yoshikawa, K.; Falk, M.; Hildenbrand, G.; Salmina, K.; Freivalds, T.; Vainshelbaum, N.; Weidner, J.; Sievers, A.; et al. Spatial-Temporal Genome Regulation in Stress-Response and Cell-Fate Change. Int. J. Mol. Sci. 2023, 24, 2658. https://doi.org/10.3390/ijms24032658
Erenpreisa J, Giuliani A, Yoshikawa K, Falk M, Hildenbrand G, Salmina K, Freivalds T, Vainshelbaum N, Weidner J, Sievers A, et al. Spatial-Temporal Genome Regulation in Stress-Response and Cell-Fate Change. International Journal of Molecular Sciences. 2023; 24(3):2658. https://doi.org/10.3390/ijms24032658
Chicago/Turabian StyleErenpreisa, Jekaterina, Alessandro Giuliani, Kenichi Yoshikawa, Martin Falk, Georg Hildenbrand, Kristine Salmina, Talivaldis Freivalds, Ninel Vainshelbaum, Jonas Weidner, Aaron Sievers, and et al. 2023. "Spatial-Temporal Genome Regulation in Stress-Response and Cell-Fate Change" International Journal of Molecular Sciences 24, no. 3: 2658. https://doi.org/10.3390/ijms24032658