
On the Intermolecular Interactions in Thiophene-Cored Single-Stacking Junctions

Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovsky Square 2, 16200 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(17), 13349; https://doi.org/10.3390/ijms241713349
Submission received: 14 August 2023
/
Revised: 23 August 2023
/
Accepted: 24 August 2023
/
Published: 28 August 2023
(This article belongs to the Special Issue Non-covalent Interaction 2.0)
Abstract
:There have been attempts, both experimental and based on density-functional theory (DFT) modeling, at understanding the factors that govern the electronic conductance behavior of single-stacking junctions formed by pi-conjugated materials in nanogaps. Here, a reliable description of relevant stacked configurations of some thiophene-cored systems is provided by means of high-level quantum chemical approaches. The minimal structures of these configurations, which are found using the dispersion-corrected DFT approach, are employed in calculations that apply the coupled cluster method with singles, doubles and perturbative triples [CCSD(T)] and extrapolations to the complete basis set (CBS) limit in order to reliably quantify the strength of intermolecular binding, while their physical origin is investigated using the DFT-based symmetry-adapted perturbation theory (SAPT) of intermolecular interactions. In particular, for symmetrized S-Tn dimers (where “S” and “T” denote a thiomethyl-containing anchor group and a thiophene segment comprising “n” units, respectively), the CCSD(T)/CBS interaction energies are found to increase linearly with n ≤ 6, and significant conformational differences between the flanking 2-thiophene group in S-T1 and S-T2 are described by the CCSD(T)/CBS and SAPT/CBS computations. These results are put into the context of previous work on charge transport properties of S-Tn and other types of supramolecular junctions.
1. Introduction
Currently, one of the key research directions in molecular electronics [1] aims at controlling charge transport properties in supramolecular junctions [2]. The most frequently studied supramolecular junctions are formed by π-conjugated molecules (see the review [3] and also the most recent papers [4,5]). Specifically, single-stacking supramolecular junctions composed of thienyl-capped oligothiophenes were carefully investigated by Hong and his coworkers (see reference [6] and work cited therein). On the experimental side, these studies achieved reliable simultaneous measurements of conductance and the electromechanical coupling factor (denoted as α, see reference [7]) in gold electrode–single supermolecule–gold electrode junctions formed in a nanogap. This way, a series of S-Tn dimers (where S and T denote a thiomethyl-containing anchor group and a thiophene segment comprising n units, respectively; the actual structures are shown in Section 2) was examined for n ranging from one to four, together with control experiments for the corresponding S-Tn-S single molecules [6]. Significantly, the stacking arrangement of S-Tn dimers in the junctions had been previously established [8], which is a crucial structural feature of these systems that is generally not present in, for instance, the iodine-terminated oligothiophenes studied by Tao et al. [9]. On the theoretical side, density-functional theory (DFT) and molecular dynamics (MD) calculations were performed for the S-Tn dimers [6]. Moreover, for the related single-stacking junctions, an important spring model was derived to analyze the strain distribution in them [6], and the charge transport characteristics of some monomers and dimers positioned in between gold electrodes were simulated [6,8]. This combination of measurements and modeling provided insights into noncovalent interactions at single-molecule level and thus led to an elucidation of specific features of conductance behavior of thienyl-capped oligothiophenes, which should be important in the field of single-supermolecule electronics [10].
In this work, the aforementioned DFT computational description of S-Tn dimers is substantially extended. The following three main questions are addressed. (1) What are the global minima of overlapping configurations of the S-Tn dimers with n ranging from one to four? In Section 2.1, these minima are compared to the corresponding structures that were considered by Hong et al. [6]. The comparison is centered on computations of intermolecular interaction energy, , employing the domain-based local pair natural orbital (DLPNO) variant of the coupled cluster theory with singles, doubles and perturbative triples [CCSD(T)] and extrapolations to the complete basis set (CBS) limit for geometries located using the dispersion-corrected DFT approach (see Section 4 for references and technical details of the resulting DLPNO-CCSD(T)/CBS//B97-D/def2-TZVPP method). Based on this comparison, in Section 2.2 the Ci symmetric geometries are chosen to investigate an extended set of pertinent structures in order to answer the second main question: (2) what is the trend in data for the S-Tn dimers comprising one to six thiophene units? It should be mentioned that predicted values of are vital for an interpretation of the measured probability of stacking in thiophene-based junctions [8]. The third main question, which is addressed in Section 2.3, is: (3) how big are conformational differences between the flanking 2-thiophene group in S-T1 and S-T2 dimers? These differences are crucial for an explanation of the “odd–even effect” that concerns distinct strains of intermolecular interactions in S-Tn dimers for odd and even values of n, as detailed in reference [6]. Hence, the rotation of terminal 2-thiophene rings is described in terms of the CCSD(T)/CBS -values of pertinent conformers. Furthermore, the DFT-based symmetry-adapted perturbation theory (SAPT) of intermolecular interactions [11] (see Section 4 for specifications) is applied to those conformers. Answers to questions (1)–(3) are discussed in Section 3 together with some thermodynamic considerations. They support the characterization provided by Hong et al. of the behavior of the S-Tn dimers in nanogaps under experimental conditions [6] and are of importance for understanding the factors that control charge transport of single-stacking junctions in general [12,13]. Moreover, the present theoretical approach can be immediately applied to other dimers in the process of modeling novel materials.
2. Results
2.1. The Energy Minima
Molecular structures of monomers forming S-Tn dimers are shown in Figure 1, while the optimized geometries of all dimers that are discussed in this work are provided, in xyz format, inside Supporting Information file “structures.tar”. For the S-Tn dimers with n ranging from one to four, the potential energy surface (PES) was scanned in a region relevant for the formation of stacked configurations. In short, numerous starting orientations were prepared, and minima of the PES were sought by the B97-D/def2-TZVPP approach (it should be noted that this method was recently used to search the PES of numerous dimers of heterocycles [14]). The resulting minima were ranked by the DLPNO-CCSD(T)/CBS estimation of the using the focal point procedure developed most recently [15]. As detailed in reference [15], this procedure was shown to provide -values accurate to within about 2 kJ/mol for some challenging cases from the L7 set [16] and for other complex systems. Here, it was validated by considering the conformers of the Ci symmetric S-T1 dimer, which are described in Section 2.3. For these conformers, the canonical CCSD(T)/CBS data were obtained (see Section 4 for details) and compared to their DLPNO-CCSD(T)/CBS counterparts. Only five geometries were considered due to an exceedingly high computational cost of the underlying canonical CCSD(T) step for this S-T1 dimer, which contains 46 atoms. Nevertheless, the maximum and mean absolute deviations of the two data sets amount to 1.62 and 1.49 kJ/mol, respectively. The interaction energies are summarized in Supporting Information Table S1 and show only a relatively small and systematic underestimation of the DLPNO-CCSD(T)/CBS relative to the canonical CCSD(T)/CBS data (raw values of all absolute energies are provided in Supporting Information spreadsheets “DLPNO.xlsx” and “canonical.xlsx”).
Importantly, the authors of reference [6] kindly sent us coordinates of their structures of S-Tn dimers for n from one to four. An inspection of the obtained structures revealed that they were all of nearly Ci symmetry. Hence, they were symmetrized and reoptimized at the B97-D/def2-TZVPP level. The resulting structures are referred to as “symmetric” from now on. Analogous Ci symmetric dimers with n of five and six are also considered in this work (see Section 2.2). The most stable structures according to the aforementioned ranking based on the DLPNO-CCSD(T)/CBS interaction energy are denoted as “unsymmetric” in Table 1, Table 2 and Table 3 and the related discussion.
Furthermore, it was checked how structural features and interaction energies would change if some method more involved than the B97-D/def2-TZVPP is employed for the geometry optimization. In the next paragraph, such results are presented for the S-T1 dimer and the double-hybrid B2PLYP-D3/def2-QZVPPD approach (see Section 4 for references and technical details), which is highly accurate [17] but computationally quite demanding because it requires the second-order Møller–Plesset (MP2) correlation energy estimation using an ample def2-QZVPPD basis set (2362 basis functions in the case of the S-T1 dimer).
Table 1 summarizes results obtained for the B97-D/def2-TZVPP and (values in parentheses) B2PLYP-D3/def2-QZVPPD minima of two types of stacked S-T1 dimers. For each structure in Table 1, the total DLPNO-CCSD(T)/CBS interaction energy, which is denoted as , is presented in terms of its three constituting parts. These parts are the Hartree–Fock (), MP2 correlation (), and higher-order correlation () energy contributions. Details of an estimation of the CBS-extrapolated , , and data are given in Section 4. Further, in Table 1, the total interaction energy obtained from the SAPT treatment, , is shown together with its breakdown into the electrostatic polarization, first-order exchange, and induction and dispersion contributions (see Section 4), which are denoted as , , , and , respectively.
As immediately follows from the and data in Table 1, the interaction energy is only slightly affected by differences in geometry caused by the choice of the optimization method (an overlay of the B97-D/def2-TZVPP and B2PLYP-D3/def2-QZVPPD structures of the unsymmetric S-T1 dimer is pictured in Supporting Materials Figure S1 together with values of some intermolecular distances). It should be also noticed that the total interaction energies agree between each other to within one kJ/mol for all related structures of the S-T1 dimer. Moreover, ratios of the respective contributions to the total interaction energy do not significantly depend on the method that was used to optimize the geometry. This holds for both the supermolecule and SAPT calculations (see Table 1). Hence, only the B97-D/def2-TZVPP geometries were considered for larger S-Tn dimers, which are discussed further on in this paper.
The two investigated types of stacked S-T1 dimers in their B97-D/def2-TZVPP minima are pictured in Figure 2. Expectedly, they can be categorized as van der Waals dispersion-dominated [18]. In the unsymmetric arrangement, the dispersion-to-electrostatic ratio (see reference [19] and Section 4), , is 2.25 according to the SAPT-DFT/CBS calculations carried out for the B97-D/def2-TZVPP geometry, and there are methyl groups positioned on top of 2-thiophene rings (see also Figure S1, and Tables S2 and S3). Such contacts are not present in the symmetric arrangement, and a partial overlap of 2-thiophene and phenyl rings is preferred instead in this case, with the corresponding of 2.65. Despite these structural dissimilarities, values of the total interaction energy differ only by a small amount between the two types of intermolecular complexes. In particular, the difference in and data amounts to 5.5 and 4.1 kJ/mol, respectively, for the B97-D/def2-TZVPP minima (see Table 1). Such small interaction energy differences indicate that there are a number of contact configurations accessible for individual supramolecular junctions at room temperature, while the presence of those configurations is of course known to affect the dispersion and shape of the α and conductance histograms [6,7,20,21].
As already explained in the preceding part, only the B97-D/def2-TZVPP-optimized structures of the S-T2 (and larger) dimers are considered here. Their interaction energies are summarized in Table 2 and demonstrate a good agreement between the and data. This shows that the SAPT-DFT/CBS results are fully reliable. They thus confirm that van der Waals dispersion dominates the binding between thiophene-based systems. Namely, for the unsymmetric and symmetric S-T2 dimer, amounts to 2.98 and 3.14, respectively (see also Table S4). Figure 3 graphically represents these structures. They feature the same intermolecular contacts as those found in the S-T1 dimers, that is, methyl/π(2-thiophene) and π(2-thiophene)/π(phenyl) interactions in the unsymmetric and symmetric structure, respectively. Differences in the total interaction energies between the two types of S-T2 dimers are also quite small (see Table 2).
In line with results described so far for the two smaller structures, differences in the total DLPNO-CCSD(T)/CBS interaction energy between the investigated configurations of S-T3 and S-T4 dimers are also relatively small (see Table 3). These larger dimers are graphically represented in Figure 4 and Figure 5. It is worth noting that the most stable unsymmetric minimum of S-T3 dimer does not accommodate methyl/π(2-thiophene) contacts, but instead it features π(2-thiophene)/π(phenyl) and π(central 2-thiophene)/π(central 2-thiophene) interactions. However, in the corresponding S-T4 dimer structure, there are methyl/π(2-thiophene) contacts present, and both its monomers have strongly bent geometries (see Figure 5). At this point, it should be mentioned that in all monomers forming symmetric dimers, the core thiophene segment is almost planar (pertinent dihedral angles have values around 20°).
2.2. The Molecular Size Dependence of
The DLPNO-CCSD(T)/CBS//B97-D/def2-TZVPP computational methodology was also applied to two larger dimers that had not been studied experimentally in reference [6], namely S-T5 and S-T6. Two important geometrical parameters of the minima of all structures are collected in Table 4. It should be noted that distances between thiomethyl anchor groups as expressed by the parameter are higher in the symmetric structures than in their unsymmetric counterparts for S-Tn dimers with n ranging from one to four. Importantly, the higher -value is, in the case of S-T1, consistent with distances estimated through conductance measurements of related single-stacking junctions (see references [6,8] for details). Hence, the PES search was not performed for S-T5 and S-T6 dimers, and only their symmetric configurations are considered here. It should also be noted that for the unsymmetric minima, the separation between centers of mass of monomers (the parameter in Table 4) decreases monotonically with the increasing size of monomers. This trend is caused by a pronounced bending of monomers in bigger unsymmetric structures, which was already mentioned in the previous paragraph.
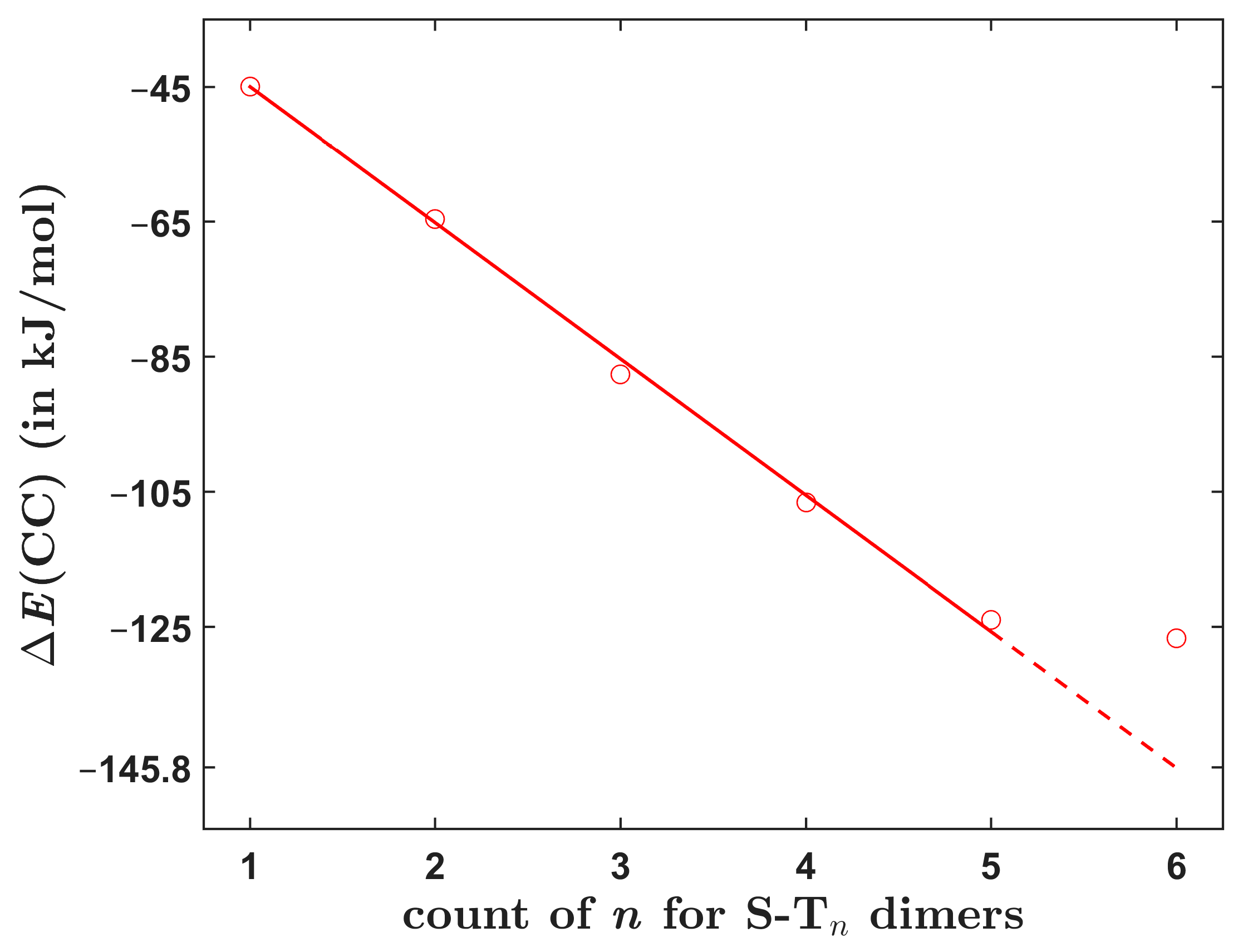
Table 5 presents the interaction energy data for S-T5 and S-T6 dimers. An inspection of their structures reveals that they contain analogous intermolecular interactions as smaller symmetric S-Tn dimers, namely π/π stacking between thiophene rings in the cores and between thiophene and phenyl rings that are located closer to terminal groups. Thus, the total interaction energy might be expected to increase strictly linearly in the whole S-Tn series, that is, for all n from one to six. Interestingly, this is the case only for n up to and including five (see Figure 6). For these five dimers, it is convenient to fit the dependence of upon n to the linear form kJ/mol, which uses an intercept fixed at the interaction energy value for S-T1. This is a robust value since it is close to the canonical CCSD(T)/CBS result of ca. −46.5 kJ/mol and also to the SAPT-DFT/CBS result of ca. −46.0 kJ/mol (see Section 2.3). The fit is highly accurate (with an average and maximum absolute residual of 1.4 and 2.3 kJ/mol, respectively, and the adjusted R2 = 0.9997). The resulting value of a slope, a = −20.19 with a standard error of 0.28, then approximates a monotonous increase of with the size of investigated dimers. Clearly, this dependence does not well describe the total interaction energy in S-T6, which is only 2.9 kJ/mol higher in absolute value than its counterpart for S-T5 (see Table 5 and Figure 6, where the dashed line is used for an extrapolation to n = 6). Analogous dependencies were obtained for the , , and data and are presented in Figures S2–S4. They show the same trend, that is, a linear growth of each term with the system size. This growth is practically uniform for the S-Tn dimers with n ≤ 5 but is much smaller for S-T6. The origin of the seemingly too small (in absolute value) of S-T6 is currently unknown. One might speculate about a big interatomic three-body dispersion contribution [22] that had perhaps strongly lowered the binding in S-T6, or even about a possible disagreement between the CCSD(T) and quantum diffusion Monte Carlo descriptions of this large (comprising 116 atoms) polarizable complex [23]. Nevertheless, it is worth noting that at the B97-D/def2-TZVPP level, the interaction energies follow a strictly linear trend for all six S-Tn dimers but are significantly overestimated relative to the data (see Figure S5 and Table S5).
2.3. The Conformational Dependence of
Various aspects of a relationship between conductance and conformational behavior of single-molecule junctions were described (see reference [24] and work cited therein and also the most recent references [25,26]). Less is known about this relationship in dimer junctions [27]. Rotations of terminal 2-thiophene rings were considered in an analysis of conductance and α-value measurements by Hong and coworkers [6]. Specifically, DFT and MD calculations were used to interpret the experimentally observed “odd-even” effect in α-values for S-Tn dimers with n ranging from one to four [6]. Here, the dependence of binding strength upon local conformational changes of flanking 2-thiophenes was investigated by means of the CCSD(T)/CBS and DFT-SAPT/CBS computations. The symmetrized B97-D/def2-TZVPP geometries of S-T1 and S-T2 dimers were employed to vary the dihedral angles that are denoted as β in the following and express a departure of the terminal 2-thiophene ring from planarity relative to the preceding ring. For these dimers, Figures S6 and S7 show chains of atoms defining the respective β angle. Due to the Ci symmetry of the dimers, their constituting monomers have a dihedral angle with a value of either +β or −β. It is noted that analogous dihedral angles were reported as (180° − β) in reference [6].
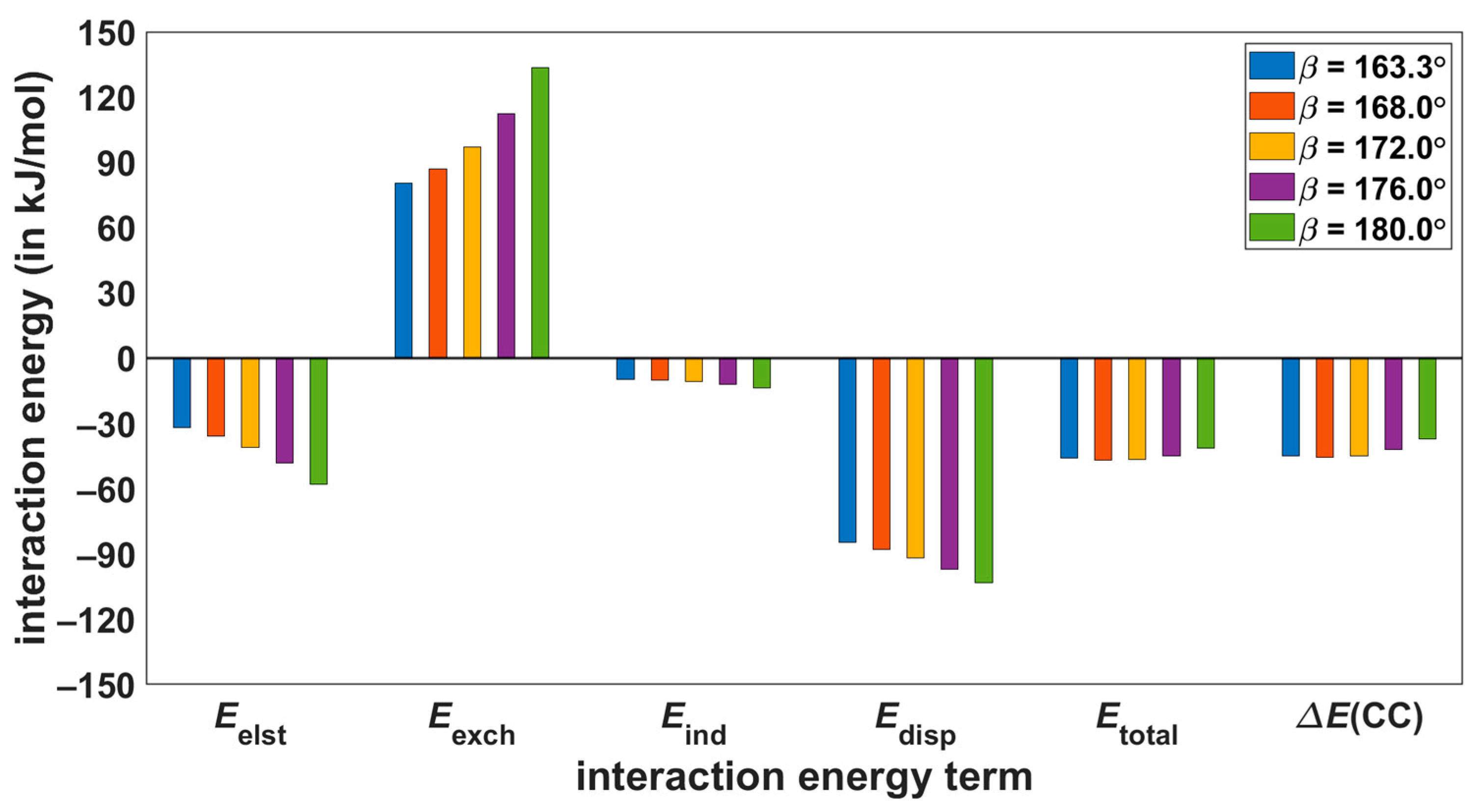
The dependence of individual interaction energies upon the angle β is graphically represented in Figure 7 and Figure 8 for S-T1 and S-T2 systems, respectively (additional details can be found in Tables S1, S6 and S7). For both dimers, structures with the lowest β-value correspond to the pertinent B97-D/def2-TZVPP minimum, and the conformational region between this value and β = 180° is sampled. Importantly, the high-level calculations show that a decrease in the absolute value of the total interaction energy with increasing value of β is about two times lower in S-T1 than in S-T2 dimers. For example, in the S-T1 dimer, the changes by |7.7| kJ/mol between structures with β of 163.3° and 180.0°, while in the S-T2 dimer, the changes by |14.7| kJ/mol between structures with β = 163.5° and 180.0°. Such variations of course indicate a higher degree of local conformation freedom in S-T1 as compared to the S-T2 dimer, in agreement with reference [6]. This trend in the data is confirmed by DFT-SAPT/CBS results. In particular, for the same example as above, the differences in are |4.6| and |13.1| kJ/mol for the corresponding β changes in the S-T1 and S-T2 dimers, respectively. It should also be mentioned that the CCSD(T) and DFT-SAPT methods place a minimum of the total interaction energy of S-T1 between the first and third (in ascending order) β-value (see Figure 7 and Table S1), while these methods agree that absolute values of the total interaction energy decrease monotonically with increasing β in the case of S-T2 dimers (see Figure 8 and Table S2). Thus, the DFT-SAPT/CBS data should be of interest as they are quite accurate and describe the physical origin of intermolecular interactions. Analysis of these data reveals that for all investigated β angles, the intermolecular binding in both the S-T1 and S-T2 dimers is dominated by van der Waals dispersion (underlying values of the interaction energy components are collected in Tables S6 and S7). Specifically, in the S-T1 system, the ratio amounts to 2.65 for β = 163.3°, and for β = 180.0° it is reduced to 1.79. This ratio equals 3.14 for β = 158.2° and drops to 2.71 for β = 180.0° in the S-T2 dimers.
3. Discussion
Here, various dimers of thiophene-cored structures were computationally studied in their stacked arrangements with the general goal of accurately describing intermolecular interactions in this type of complex. Importantly, in reference [6], stacked configurations of the four smaller oligothiophene systems were characterized by the mechanical and conductance measurements of dimer junctions; an “odd-even” trend in α-values was found. This trend, which represents differences in the strain distribution of intermolecular interactions of single-stacking junctions, was then interpreted using DFT and MD modeling [6]. Results obtained in the present work provide an extensive description of structures and intermolecular binding of related S-Tn dimers with n ranging from one to six. Several specific questions about these adducts, which were posed in the Introduction, are discussed in this section.
The PES search was performed at the DLPNO-CCSD(T)/CBS//B97-D/def2-TZVPP level for the S-Tn complexes with n ≤ 4 in their overlapping configurations. It unveiled the minima that were more strongly bound than their counterparts of the Ci molecular symmetry. Interestingly, all these enthalpically more favorable structures feature significantly shorter distances between thiomethyl anchors than the corresponding Ci symmetric geometries do. However, it appeared that geometries with longer -values would be more representative of the junctions investigated experimentally [6,8], and only the symmetric geometries were considered while following structure dependencies that are described below.
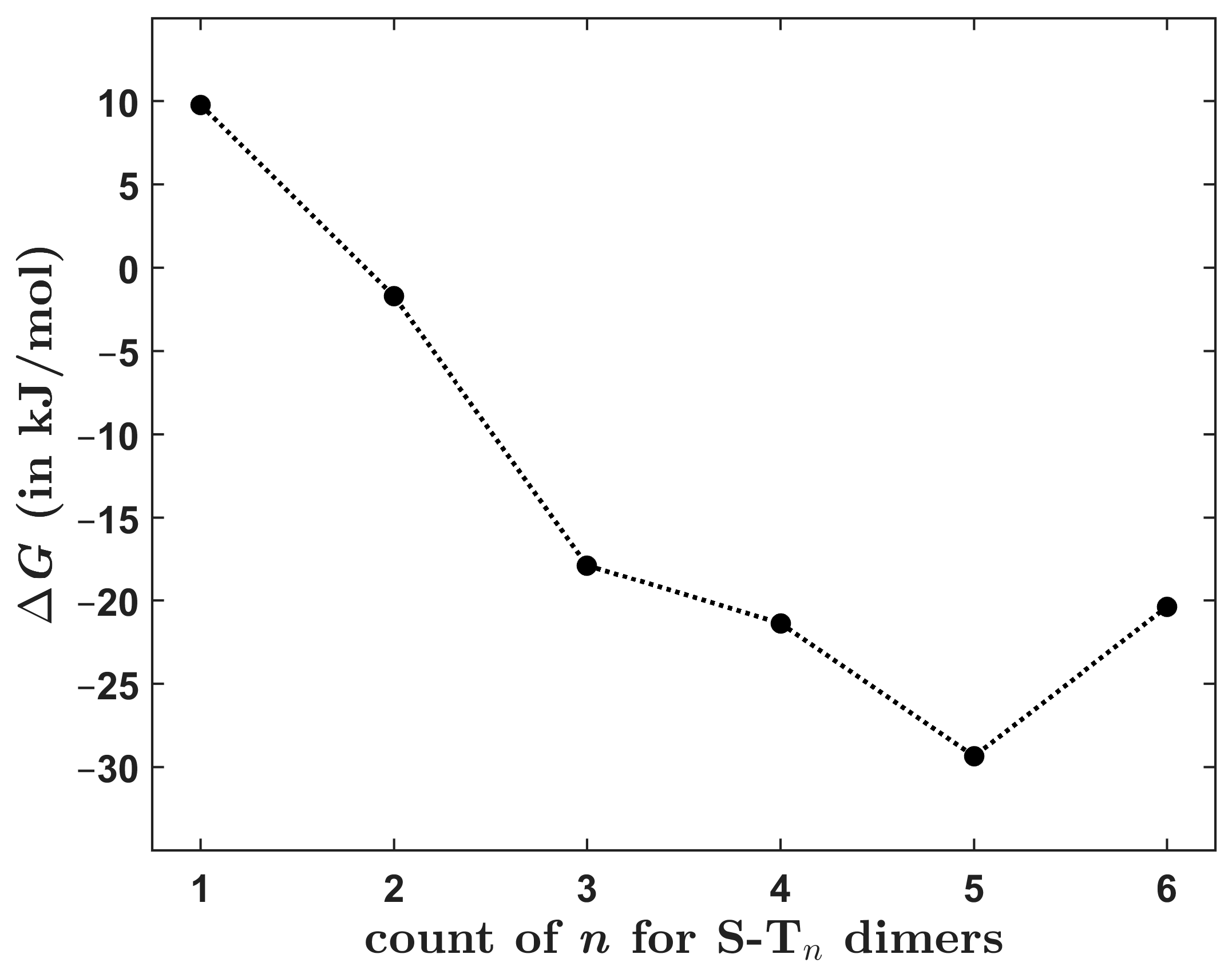
The dependence of the interaction energy upon the molecular size was inspected for the six symmetric dimers. Absolute values of were found to monotonously increase with the system size. This rise was fairly large and almost uniform (20.19 ± 0.28 kJ/mol according to the linear model specified in Section 2.2) for the S-Tn dimers with n ≤ 5 but was only negligible (amounting to 2.9 kJ/mol) for the biggest system, that is, S-T6. Hence, it may be worthwhile to also predict a change in the Gibbs free energy accompanying the formation of dimers under standard thermodynamic conditions, . Only a crude method for the estimation was used, which employed the DLPNO-CCSD(T)/CBS interaction energies together with results of the B97-D/def2-TZVPP harmonic vibrational analysis of each dimer and its constituting monomers. Then, for the assumed dimerization process at the given temperature, related changes in the zero-point vibrational energies, the vibrational thermal energies, and the entropies were calculated routinely [28] and were combined with the data to arrive at a value for the Gibbs free energy of formation. Results obtained for a temperature of 298.15 K are graphically represented in Figure 9 (underlying values of the thermodynamic parameters are collected in Table S8). They show a complicated behavior of as a function of molecular size. Namely, a positive -value is predicted for S-T1, in contrast to results obtained for the rest of the investigated dimers. Furthermore, the of S-T4 is slightly higher in absolute value than its counterpart for S-T6. This demonstrates that there can be “sweet spots” of thermodynamic stabilization of stacked dimers for certain sizes of constituting monomers. In the present case, such a “sweet spot” was apparently found at n = 5 (see Figure 9).
Moreover, dependencies of the interaction energy upon varying dihedral angles, which define orientations of terminal 2-thiophene rings with respect to the core of S-T1 and S-T2 dimers, were followed. It was suggested in reference [6] that differences in rotational barriers of these rings in single-stacking oligothiophene junctions led to the “odd-even” effect in the measured α-values. In this work, the rings were rotated between their position in the B97-D/def2-TZVPP minimum and their planarity relative to the rest of the dimer. For the rotated structures, the CCSD(T)/CBS and SAPT-DFT/CBS interaction energies were obtained. These calculations revealed much lower changes in the interaction energy with the rotation in S-T1 as compared to the S-T2 dimer. This result is consistent with a higher conformational freedom in the smaller system and is thus in line with an interpretation of the “odd-even” effect provided in reference [6].
4. Materials and Methods
Geometry optimizations and subsequent estimations of harmonic vibrational frequencies and intermolecular interaction energies were performed using the Gaussian 16, revision C.01 suite of codes [29] with default settings. These computations used the B97-D/def2-TZVPP (the B97 functional [30] combined with the empirical dispersion correction from reference [31] and applied together with the TZVPP basis set [32]) and the B2PLYP-D3/def2-QZVPD (the double-hybrid B2-PLYP functional [33,34] combined with the D3 empirical dispersion correction [35] and applied together with the QZVPPD basis set [36]; see also reference [37]) approaches.
The canonical CCSD(T)/CBS interaction energies were obtained by the focal-point method expressed by Equation (1) (see reference [15] for further details):
where subscripts denote the respective energy terms, namely the total Hartree–Fock energy (HF), the MP2 correlation energy (MP2), and the higher-order correlation energy (post-MP2), and superscripts specify the basis set used to compute the respective term. Abbreviations “aTZ”, “aQZ”, and “a5Z” denote the standard augmented correlation-consistent polarized valence triple-zeta, quadruple-zeta, and quintuple-zeta basis sets, respectively [38,39]. The MP2/a5Z correlation energies were obtained in the resolution-of-the-identity integral approximation [40,41] while using the relevant auxiliary basis sets [41]. Calculations of the HF/a5Z and MP2/a5Z energies were performed in Turbomole, version 7.1 [42]. Calculations of the canonical CCSD(T)/aTZ and MP2/aTZ correlation energies were carried out in Molpro 2021.2 [43].
The SAPT-DFT/CBS interaction energies were estimated using the same procedures as in our most recent work [44]. The , , , and contributions to the total interaction energy, , from Section 2 are related to the underlying interaction energy terms as follows: and are the polarization and exchange energy contributions, respectively, arising in the first order of the perturbation theory of intermolecular interactions [45]; is the dispersion energy contribution obtained as a sum of the second order terms and [46]; and is the induction energy contribution approximated by a sum of the second order terms and [47] and the correction term , which is computed at the HF level [48]. All the related calculations were performed in Molpro 2021.2.
The DLPNO-CCSD(T)/CBS interaction energies were approximated by the focal-point method from reference [15], which applies Equation (2) (the notation is as in Equation (1), and a right arrow is used to indicate an application of the two-point extrapolation formula from reference [49]):
while the CCSD(T) and MP2 correlation energies were obtained in the DLPNO approximation [50,51,52,53]. The ORCA 5.0.3 program package [54] was used with the “TightPNO” set of parameters for the truncation of the electron-correlation space and with the default method of orbital localization.
5. Conclusions
The main findings of this work can be summarized according to the three key questions that were posed in the Introduction, as follows. Firstly, -values obtained for global minima of overlapping configurations of the S-Tn dimers with n ranging from one to four are only slightly higher than for the corresponding Ci symmetric structures. Secondly, -values grow linearly with the system size for the S-Tn dimers with n up to and including six. Thirdly, in agreement with an interpretation of the “odd-even” effect that was found experimentally in the junctions [6], a hindrance of the rotation of terminal 2-thiophene rings is much higher in S-T2 than in S-T1. In addition, the presented DFT-SAPT analysis of intermolecular interactions highlights the dominant role of van der Waals dispersion in the stabilization of thiophene-cored dimers. These results are expected to be of interest in the computational design of novel materials.
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms241713349/s1.
Author Contributions
Conceptualization, J.C. and J.B.; investigation, J.C.; writing, J.C.; validation, J.B.; funding acquisition, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
The Czech Science Foundation (grant GA 20-01233S).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and in the Supplementary Materials.
Acknowledgments
Computational resources were provided by the e-INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic, and by the ELIXIR-CZ project (ID:90255), part of the international ELIXIR infrastructure. We are grateful to the authors of reference [6] for sending us the Cartesian coordinates of four structures (see Section 2.1 of this paper) and also for answering our queries about their work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, T.; Bandari, V.K.; Schmid, O.G. Molecular Electronics: Creating and Bridging Molecular Junctions and Promoting Its Commercialization. Adv. Mater. 2023, 35, 2209088. [Google Scholar] [CrossRef]
- Li, X.; Ge, W.; Guo, S.; Bai, J.; Hong, W. Characterization and Application of Supramolecular Junctions. Angew. Chem. 2023, 62, 202216819. [Google Scholar] [CrossRef]
- Ayinla, R.T.; Shiri, M.; Song, B.; Gangishetty, M.; Wang, K. The pivotal role of non-covalent interactions in single-molecule charge transport. Mater. Chem. Front. 2023, 7, 3524–3542. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, J.; Wu, Q.; Hou, S.; Feng, S.; Jiang, B.; Lambert, C.J.; Gao, X.; Li, Y.; Li, J. Enhanced π–π Stacking between Dipole-Bearing Single Molecules Revealed by Conductance Measurement. J. Am. Chem. Soc. 2023, 145, 1617–1630. [Google Scholar] [CrossRef]
- Homma, K.; Kaneko, S.; Tsukagoshi, K.; Nishino, T. Intermolecular and Electrode-Molecule Bonding in a Single Dimer Junction of Naphthalenethiol as Revealed by Surface-Enhanced Raman Scattering Combined with Transport Measurements. J. Am. Chem. Soc. 2023, 145, 15788–15795. [Google Scholar] [CrossRef]
- Li, R.; Zhou, Y.; Ge, W.; Zheng, J.; Zhu, Y.; Bai, J.; Li, X.; Lin, L.; Duan, H.; Shi, J.; et al. Strain of Supramolecular Interactions in Single-Stacking Junctions. Angew. Chem. 2022, 61, e202200191. [Google Scholar] [CrossRef]
- Hihath, J.; Arroyo, C.R.; Rubio-Bollinger, G.; Tao, N.; Agraït, N. Study of Electron—Phonon Interactions in a Single Molecule Covalently Connected to Two Electrodes. Nano Lett. 2008, 8, 1673–1678. [Google Scholar] [CrossRef]
- Li, X.; Wu, Q.; Bai, J.; Hou, S.; Jiang, W.; Tang, C.; Song, H.; Huang, X.; Zheng, J.; Yang, Y.; et al. Structure-Independent Conductance of Thiophene-Based Single-Stacking Junctions. Angew. Chem. 2020, 8, 3280–3286. [Google Scholar] [CrossRef]
- Xiang, L.; Hines, T.; Palma, J.L.; Lu, X.; Mujica, V.; Ratner, M.A.; Zhou, G.; Tao, N. Non-exponential Length Dependence of Conductance in Iodide-Terminated Oligothiophene Single-Molecule Tunneling Junctions. J. Am. Chem. Soc. 2016, 138, 679–687. [Google Scholar] [CrossRef]
- Chen, H.; Stoddart, J.F. From molecular to supramolecular electronics. Nat. Rev. Mater. 2021, 6, 804–828. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Xie, X.; Li, P.; Xu, Y.; Zhou, L.; Yan, Y.; Xie, L.; Jia, C.; Guo, X. Single-Molecule Junction: A Reliable Platform for Monitoring Molecular Physical and Chemical Processes. ACS Nano 2022, 16, 3476–3505. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, Y.; Zhou, D.; Chen, Y.; Xiao, Z.; Shi, J.; Liu, J.; Hong, W. Electric Field-Induced Assembly in Single-Stacking Terphenyl Junctions. J. Am. Chem. Soc. 2020, 142, 19101–19109. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Doney, A.C.; Wheeler, S.E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2019, 141, 11027–11035. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J.; Czerneková, V. A Cost Effective Scheme for the Highly Accurate Description of Intermolecular Binding in Large Complexes. Int. J. Mol. Sci. 2022, 23, 15773. [Google Scholar] [CrossRef]
- Sedlak, R.; Janowski, T.; Pitoňák, M.; Řezáč, J.; Pulay, P.; Hobza, P. Accuracy of Quantum Chemical Methods for Large Noncovalent Complexes. J. Chem. Theory Comput. 2013, 9, 3364–3374. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J.; Czerneková, V.; Kobera, L. Quantifying the Intrinsic Strength of C–H⋯O Intermolecular Interactions. Molecules 2023, 28, 4478. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. Chem. Rev. 2016, 116, 5038–5071. [Google Scholar] [CrossRef]
- Řezáč, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef]
- Li, Z.; Mejía, L.; Marrs, J.; Jeong, H.; Hihath, J.; Franco, I. Understanding. the Conductance Dispersion of Single-Molecule Junctions. J. Phys. Chem. C 2021, 125, 3406–3414. [Google Scholar] [CrossRef]
- Mejía, L.; Kleinekathöfer, U.; Franco, I. Coherent and incoherent contributions to molecular electron transport. J. Chem. Phys. 2022, 156, 094302. [Google Scholar] [CrossRef]
- von Lilienfeld, O.A.; Tkatchenko, A. Two- and three-body interatomic dispersion energy contributions to binding in mole-cules and solids. J. Chem. Phys. 2010, 132, 234109. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamdani, Y.S.; Nagy, P.R.; Zen, A.; Barton, D.; Kállay, M.; Bradenburg, J.G.; Tchatkenko, A. Interactions between large molecules pose a puzzle for reference quantum mechanical methods. Nat. Commun. 2021, 12, 3927. [Google Scholar] [CrossRef] [PubMed]
- Mejía, L.; Renaud, N.; Franco, I. Signatures of Conformational Dynamics and Electrode-Molecule Interactions in the Conductance Profile During Pulling of Single-Molecule Junctions. J. Phys. Chem. Lett. 2018, 9, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Bates, D.; Sangtarash, S.; Ferri, N.; Thomas, A.; Higgins, S.J.; Robertson, C.M.; Nichols, R.J.; Sadeghi, H.; Vezzoli, A. Folding a Single-Molecule Junction. Nano Lett. 2020, 20, 7980–7986. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, Y.; Ren, L.; Ye, J.; Wang, H.; Liu, X.; Huang, R.; Liu, H.; Liu, J.; Shi, J.; et al. Switching Quantum Interference in Single-Molecule Junctions by Mechanical Tuning. Angew. Chem. 2023, 62, e202302693. [Google Scholar] [CrossRef]
- Magyarkuti, A.; Adak, O.; Halbritter, A.; Venkataraman, L. Electronic and mechanical characteristics of stacked dimer molecular junctions. Nanoscale 2018, 10, 3562–3568. [Google Scholar] [CrossRef]
- Irikura, K.K.; National Institute of Standards and Technology. Using the Output File from a Gaussian Frequency Calculation to Compute Ideal-Gas Thermodynamic Functions. Available online: https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas/ (accessed on 12 August 2023).
- Frish, M.J.; Trucks, J.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Becke, A. Density-Functional Thermochemistry. V. Systematic Optimization of Exchange-Correlation Functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Brémond, É.; Savarese, M.; Su, N.Q.; Pérez-Jiménez, Á.J.; Xu, X.; Sancho-García, J.C.; Adamo, C. Benchmarking Density Functionals on Structural Parameters of Small-/Medium-Sized Organic Molecules. J. Chem. Theory Comput. 2016, 12, 459–465. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simula-tions. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Hesselmann, A.; Kats, D.; Kohn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [PubMed]
- Czernek, J.; Brus, J.; Czerneková, V. A computational inspection of the dissociation energy of mid-sized organic dimers. J. Chem. Phys. 2022, 156, 204303. [Google Scholar] [CrossRef] [PubMed]
- Heßelmann, A.; Jansen, G. First-order intermolecular interaction energies from Kohn—Sham orbitals. Chem. Phys. Lett. 2002, 357, 464–470. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. Intermolecular dispersion energies from time-dependent density functional theory. Chem. Phys. Lett. 2003, 367, 778–784. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. Intermolecular induction and exchange-induction energies from coupled-perturbed Kohn—Sham density functional theory. Chem. Phys. Lett. 2002, 362, 319–325. [Google Scholar] [CrossRef]
- Moszynski, R.; Heijmen, T.G.A.; Jeziorski, B. Symmetry-adapted perturbation theory for the calculation of Hartree—Fock interaction energies. Mol. Phys. 1996, 88, 741–758. [Google Scholar] [CrossRef]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-set convergence in correlated calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef]
- Pinski, P.; Riplinger, C.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. I. An efficient and simple linear scaling local MP2 method that uses an intermediate basis of pair natural orbitals. J. Chem. Phys. 2015, 143, 034108. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
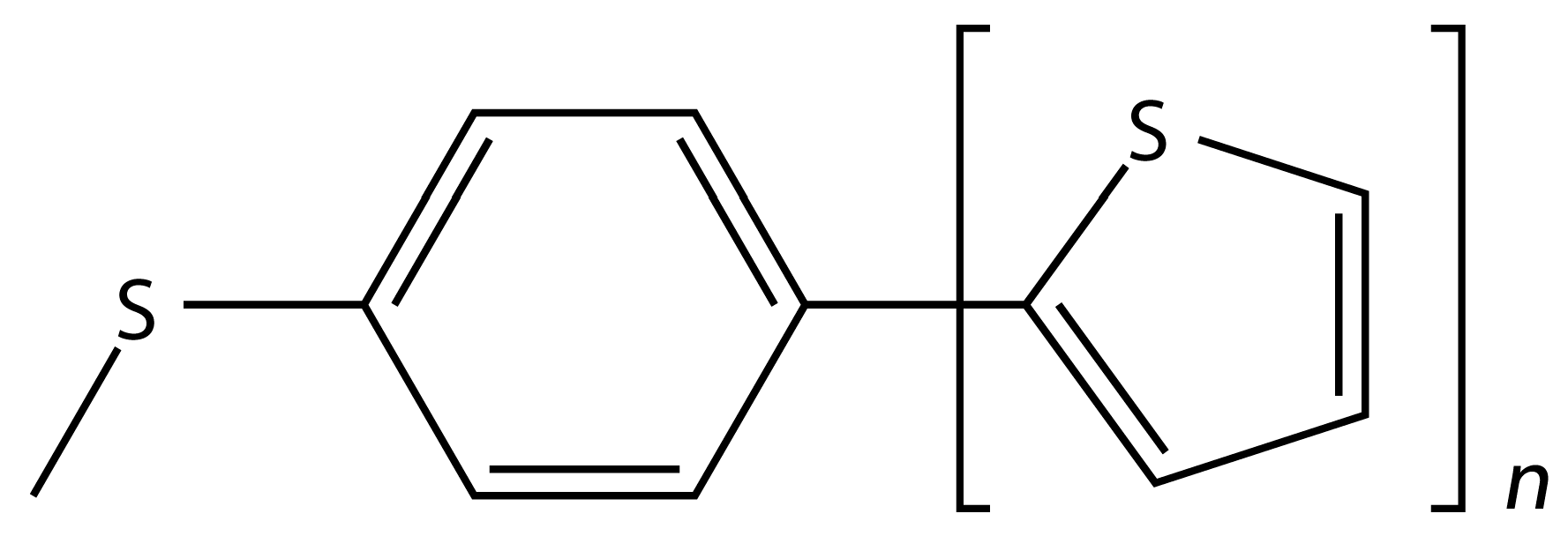
Figure 1.
Chemical structure of the building blocks of stacked dimers described in the text.

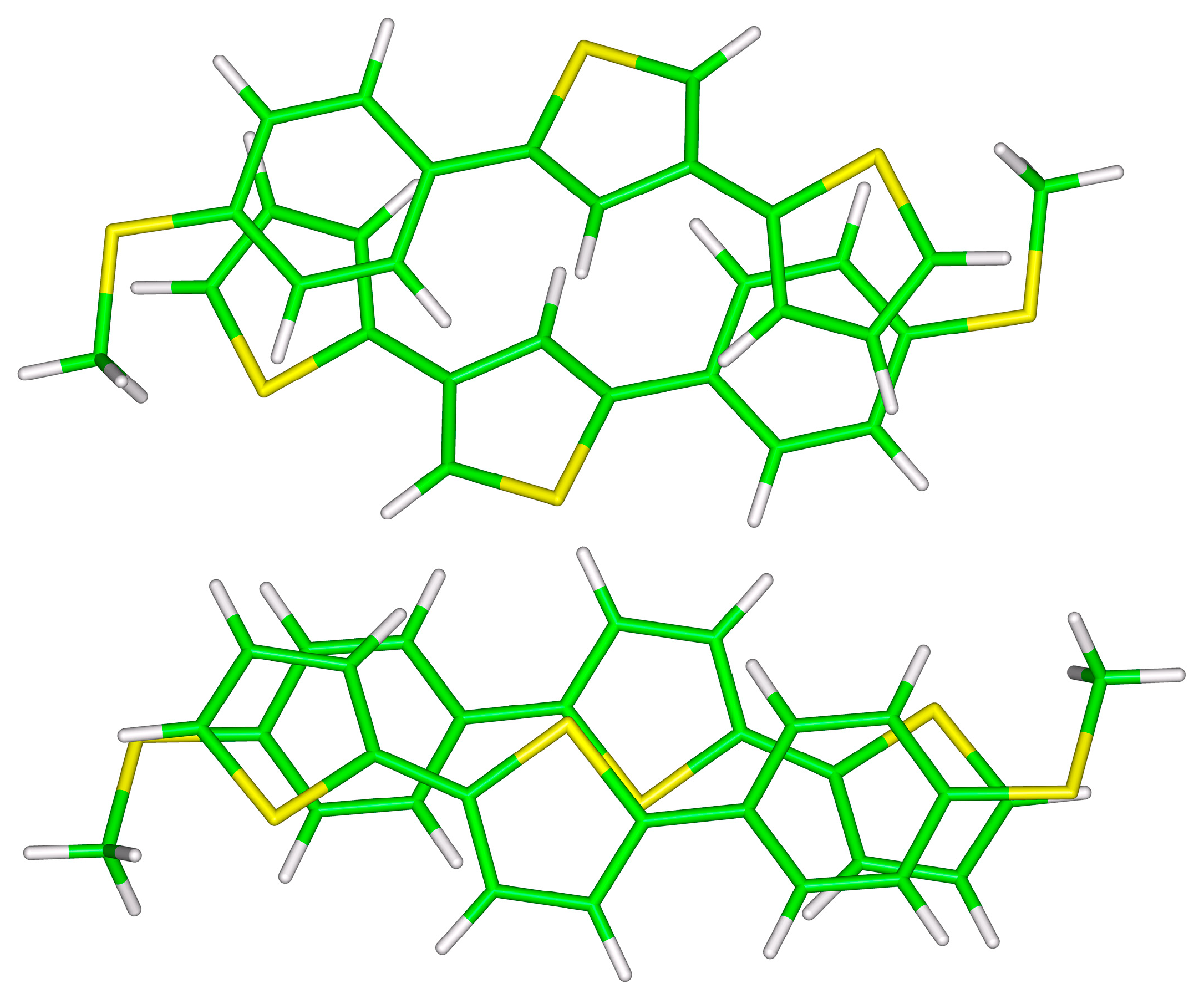
Figure 2.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T1 dimers.
Figure 2.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T1 dimers.

Figure 3.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T2 dimers.
Figure 3.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T2 dimers.

Figure 4.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T3 dimers.
Figure 4.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T3 dimers.

Figure 5.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T4 dimers.
Figure 5.
The B97-D/def2-TZVPP minima of unsymmetric (top panel) and symmetric (bottom panel) S-T4 dimers.

Figure 6.
The dependence of the total interaction energy upon molecular size of investigated dimers. The red line is specified in the text.
Figure 6.
The dependence of the total interaction energy upon molecular size of investigated dimers. The red line is specified in the text.

Figure 7.
Values extrapolated to the complete basis set limit of the intermolecular interaction energy terms for S-T1 structures with varying dihedral angle β.
Figure 7.
Values extrapolated to the complete basis set limit of the intermolecular interaction energy terms for S-T1 structures with varying dihedral angle β.

Figure 8.
Values extrapolated to the complete basis set limit of the intermolecular interaction energy terms for S-T2 structures with varying dihedral angle β.
Figure 8.
Values extrapolated to the complete basis set limit of the intermolecular interaction energy terms for S-T2 structures with varying dihedral angle β.

Figure 9.
The theoretically estimated Gibbs free energy change at 298.15 K of the formation of investigated dimers plotted against their size. Data points are connected by the dotted line.
Figure 9.
The theoretically estimated Gibbs free energy change at 298.15 K of the formation of investigated dimers plotted against their size. Data points are connected by the dotted line.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for the S-T1 dimers. All values are in kJ/mol.
Table 1.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for the S-T1 dimers. All values are in kJ/mol.
| Configuration | Supermolecular | SAPT-DFT/CBS | |||||||
|---|---|---|---|---|---|---|---|---|---|
| unsymmetric | 29.7 (25.5) | −105.2 (−99.4) | 25.1 (23.6) | −50.5 (−50.4) | −38.9 (−33.0) | 87.1 (74.7) | −10.9 (−9.5) | −87.5 (−82.5) | −50.1 (−50.2) |
| symmetric | 35.3 (33.5) | −107.5 (−105.9) | 27.2 (26.7) | −45.0 (−45.7) | −32.1 (−31.2) | 80.6 (77.6) | −9.7 (−9.0) | −84.8 (−83.8) | −46.0 (−46.3) |
Table 2.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for the S-T2 dimers. All values are in kJ/mol.
Table 2.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for the S-T2 dimers. All values are in kJ/mol.
| Configuration | Supramolecular | SAPT-DFT/CBS | |||||||
|---|---|---|---|---|---|---|---|---|---|
| unsymmetric | 51.6 | −160.7 | 41.9 | −67.2 | −41.9 | 113.6 | −12.7 | −124.9 | −65.9 |
| symmetric | 52.3 | −159.7 | 42.7 | −64.6 | −38.8 | 106.9 | −10.5 | −121.8 | −64.2 |
Table 3.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for the S-T3 and (in parentheses) S-T4 dimers. All values are in kJ/mol.
Table 3.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for the S-T3 and (in parentheses) S-T4 dimers. All values are in kJ/mol.
| Configuration | ||||
|---|---|---|---|---|
| unsymmetric | 71.1 (74.8) | −224.8 (−259.6) | 61.0 (68.5) | −92.8 (−116.3) |
| symmetric | 68.3 (81.3) | −215.0 (−261.6) | 59.2 (74.6) | −87.6 (−105.8) |
Table 4.
Selected distances in the symmetric and (in parentheses) unsymmetric dimers; ( and denote separations between centroids and sulfur atoms of thiomethyl groups of constituting monomers, respectively. All values are in pm.
Table 4.
Selected distances in the symmetric and (in parentheses) unsymmetric dimers; ( and denote separations between centroids and sulfur atoms of thiomethyl groups of constituting monomers, respectively. All values are in pm.
| System | (cc) | (SS) |
|---|---|---|
| S-T1 | 395 (399) | 1025 (834) |
| S-T2 | 380 (359) | 1396 (1225) |
| S-T3 | 391 (297) | 1784 (1538) |
| S-T4 | 400 (241) | 2189 (1724) |
| S-T5 | 402 | 2260 |
| S-T6 | 445 | 2585 |
Table 5.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for S-T5 and S-T6 dimers. All values are in kJ/mol.
Table 5.
The intermolecular interaction energy components as obtained by computational procedures specified in the text for S-T5 and S-T6 dimers. All values are in kJ/mol.
| System (in Ci Symmetry) | (CC) | |||
|---|---|---|---|---|
| S-T5 | 101.2 | −314.1 | 89.0 | −123.9 |
| S-T6 | 108.1 | −329.3 | 94.4 | −126.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Czernek, J.; Brus, J. On the Intermolecular Interactions in Thiophene-Cored Single-Stacking Junctions. Int. J. Mol. Sci. 2023, 24, 13349. https://doi.org/10.3390/ijms241713349
AMA Style
Czernek J, Brus J. On the Intermolecular Interactions in Thiophene-Cored Single-Stacking Junctions. International Journal of Molecular Sciences. 2023; 24(17):13349. https://doi.org/10.3390/ijms241713349
Chicago/Turabian StyleCzernek, Jiří, and Jiří Brus. 2023. "On the Intermolecular Interactions in Thiophene-Cored Single-Stacking Junctions" International Journal of Molecular Sciences 24, no. 17: 13349. https://doi.org/10.3390/ijms241713349
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.