
2-Acetamido-2-deoxy-d-glucono-1,5-lactone Sulfonylhydrazones: Synthesis and Evaluation as Inhibitors of Human OGA and HexB Enzymes


 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Inhibitors
2.2. Inhibition of hOGA and hHexB by 2-Acetamido-2-deoxy-d-glucono-1,5-lactone Arenesulfonylhydrazones
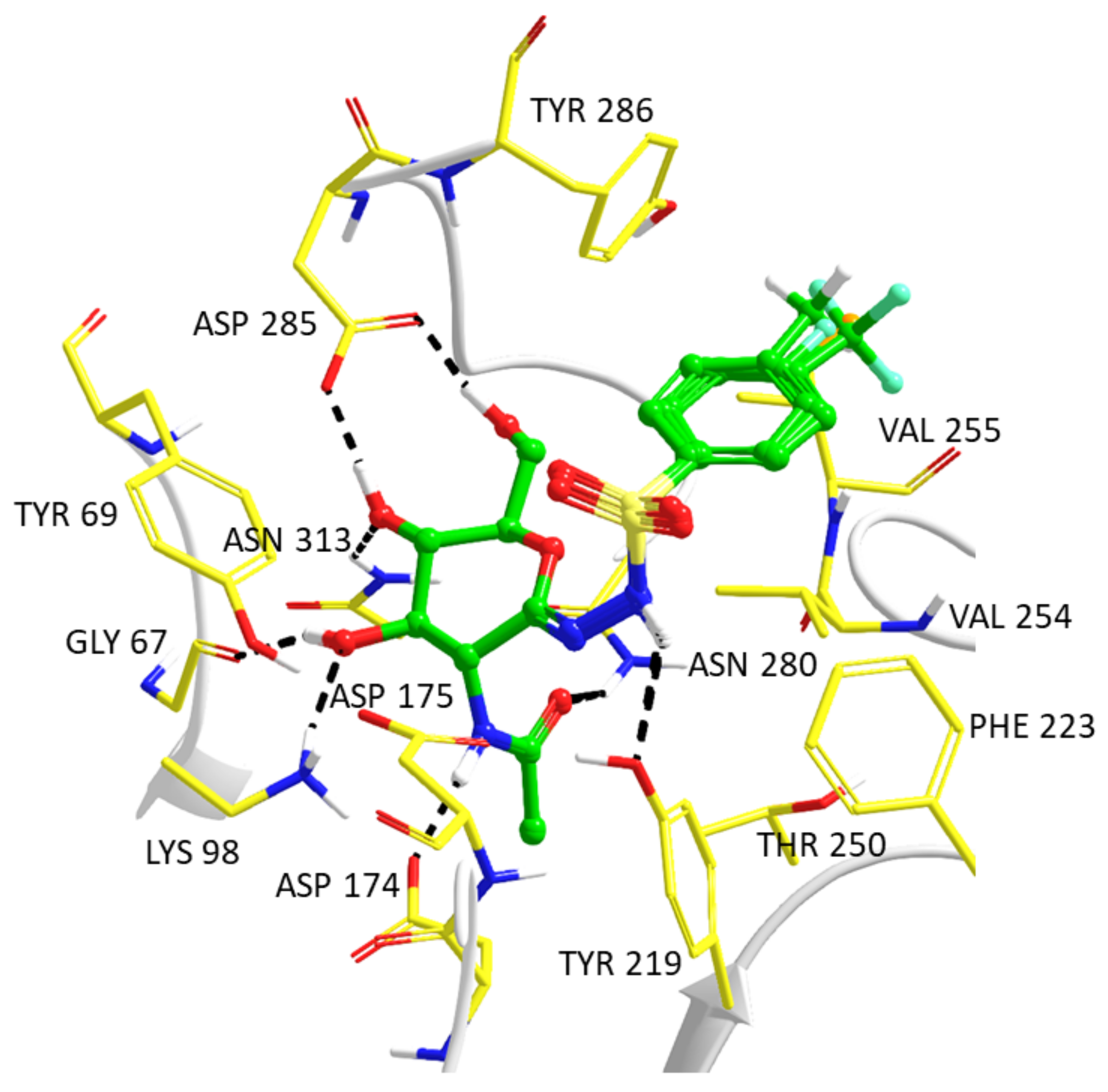
2.3. Computationally Predicted Enzyme–Inhibitor Binding in hOGA
3. Materials and Methods
3.1. Synthesis
3.1.1. General Methods
3.1.2. General Procedure A for the Synthesis of 1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-arenesulfonyl Hydrazines (3)
1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-(p-toluenesulfonyl) Hydrazine (3a)
1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-(p-trifluoromethylbenzenesulfonyl) Hydrazine (3b)
1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-(p-fluorobenzenesulfonyl) Hydrazine (3c)
1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-(p-chlorobenzenesulfonyl) Hydrazine (3d)
1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-(2-naphthalenesulfonyl) Hydrazine (3e)
1-(2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-d-glucopyranosyl)-2-(1-naphthalenesulfonyl) Hydrazine (3f)
3.1.3. General Procedure B for the Synthesis of 2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone Arenesulfonylhydrazones (4)
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone p-Toluenesulfonylhydrazone (4a)
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone p-Trifluoromethylbenzenesulfonylhydrazone (4b)
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone p-Fluorobenzenesulfonylhydrazone (4c)
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone p-Chlorobenzenesulfonylhydrazone (4d)
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone 2-Naphthalenesulfonylhydrazone (4e)
2-Acetamido-3,4,6-tri-O-acetyl-2-deoxy-d-glucono-1,5-lactone 1-Naphthalenesulfonylhydrazone (4f)
3.1.4. General Procedure C for the Synthesis of 2-Acetamido-2-deoxy-d-glucono-1,5-lactone Arenesulfonylhydrazones (5)
2-Acetamido-2-deoxy-d-glucono-1,5-lactone p-Toluenesulfonylhydrazone (5a)
2-Acetamido-2-deoxy-d-glucono-1,5-lactone p-Trifluoromethylbenzenesulfonylhydrazone (5b)
2-Acetamido-2-deoxy-d-glucono-1,5-lactone p-Fluorobenzenesulfonylhydrazone (5c)
2-Acetamido-2-deoxy-d-glucono-1,5-lactone p-Chlorobenzenesulfonylhydrazone (5d)
2-Acetamido-2-deoxy-d-glucono-1,5-lactone 2-Naphthalenesulfonylhydrazone (5e)
2-Acetamido-2-deoxy-d-glucono-1,5-lactone 1-Naphthalenesulfonylhydrazone (5f)
3.2. Biochemical Materials and Methods
3.2.1. Protein Expression and Purification
3.2.2. Inhibition Studies
3.3. Computational Details
3.3.1. Protein Preparation
3.3.2. Enzyme–Inhibitor Complex Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, J.; Wu, C.; Hart, G.W. Analytical and Biochemical Perspectives of Protein O-GlcNAcylation. Chem. Rev. 2021, 121, 1513–1581. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.R.; Hanover, J.A. O-GlcNAc Cycling: A Link between Metabolism and Chronic Disease. Annu. Rev. Nutr. 2013, 33, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Zachara, E.N. O-GlcNAc a sensor of cellular state: The role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta BBA Gen. Subj. 2004, 1673, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Hastings, N.B.; Wang, X.; Song, L.; Butts, B.D.; Grotz, D.; Hargreaves, R.; Hess, J.F.; Hong, K.-L.K.; Huang, C.R.-R.; Hyde, L.; et al. Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol. Neurodegener. 2017, 12, 1–16. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The Emerging Link between O-GlcNAc and Alzheimer Disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Zhang, L.; Li, X.; Run, X.; Liang, Z.; Li, Y.; Liu, Y.; Lee, M.H.; Grundke-Iqbal, I.; Iqbal, K.; et al. Differential Effects of an O-GlcNAcase Inhibitor on Tau Phosphorylation. PLoS ONE 2012, 7, e35277. [Google Scholar] [CrossRef]
- Yuzwa, A.S.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Dong, D.; Hart, G. Purification and characterization of an O-GlcNAc selective N-acetyl-β-d-glucosaminidase from rat spleen cytosol. J. Biol. Chem. 1994, 269, 19321–19330. [Google Scholar] [CrossRef]
- Hattie, M.; Cekic, N.; Debowski, A.W.; Vocadlo, D.J.; Stubbs, K.A. Modifying the phenyl group of PUGNAc: Reactivity tuning to deliver selective inhibitors for N-acetyl-d-glucosaminidases. Org. Biomol. Chem. 2016, 14, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Macauley, M.; Whitworth, G.E.; Debowski, A.W.; Chin, D.; Vocadlo, D. O-GlcNAcase Uses Substrate-assisted Catalysis: Kinetic analysis and development of highly selective mechanism-inspired inhibitors. J. Biol. Chem. 2005, 280, 25313–25322. [Google Scholar] [CrossRef] [Green Version]
- Macauley, M.S.; Vocadlo, D.J. Increasing O-GlcNAc levels: An overview of small-molecule inhibitors of O-GlcNAcase. Biochim. Biophys. Acta BBA Gen. Subj. 2010, 1800, 107–121. [Google Scholar] [CrossRef]
- Kiss, M.; Szabó, E.; Bocska, B.; Sinh, L.T.; Fernandes, C.P.; Timári, I.; Hayes, J.M.; Somsák, L.; Barna, T. Nanomolar inhibition of human OGA by 2-acetamido-2-deoxy-d-glucono-1,5-lactone semicarbazone derivatives. Eur. J. Med. Chem. 2021, 223, 113649. [Google Scholar] [CrossRef]
- Mangholz, S.E.; Vasella, A. Glycosylidene Carbenes. Part 5. Synthesis of Glycono-1,5-lactone Tosylhydrazones as Precursors of Glycosylidene Carbenes. Helv. Chim. Acta 1991, 74, 2100–2111. [Google Scholar] [CrossRef]
- Mangholz, S.E.; Briner, K.; Bernet, B.; Vasella, A. Glycosylidene Carbenes. Part 32. Helv. Chim. Acta 2003, 86, 2490–2508. [Google Scholar] [CrossRef]
- Somsák, L.; Praly, J.-P.; Descotes, G. Sodium Salt of d-Glucono-1,5-lactone Tosylhydrazone: A Ready Access to a New Glucosylidene Carbene Precursor. Synlett 1992, 1992, 119–120. [Google Scholar] [CrossRef]
- Chaplin, D.; Crout, D.H.G.; Bornemann, S.; Hutchinson, D.W.; Khan, R. Conversion of 2-acetamido-2-deoxy-β-d-glucopyranose (N-acetylglucosamine) into 2-acetamido-2-deoxy-β-d-galactopyranose (N-acetylgalactosamine) using a biotransformation to generate a selectively deprotected substrate for SN2 inversion. J. Chem. Soc. Perkin Trans. 1992, 1, 235–237. [Google Scholar] [CrossRef]
- Kartha, K.R.; Mukhopadhyay, B.; Field, R. Practical de-O-acylation reactions promoted by molecular sieves. Carbohydr. Res. 2004, 339, 729–732. [Google Scholar] [CrossRef]
- Zhang, G.; Fan, Q.; Wang, H.; Zhao, Y.; Ding, C. NaHSO3-Mediated Direct Synthesis of Sulfinic Esters from Sulfonyl Hydrazides under Transition-Metal-Free Conditions. Adv. Synth. Catal. 2021, 363, 833–837. [Google Scholar] [CrossRef]
- Thiele, C.M.; Petzold, K.; Schleucher, J. Easy Roesy: Reliable Cross-Peak Integration in Adiabatic Symmetrized ROESY. Chem. Eur. J. 2009, 15, 585–588. [Google Scholar] [CrossRef]
- Krejzová, J.; Kulik, N.; Slámová, K.; Křen, V. Expression of human β-N-acetylhexosaminidase B in yeast eases the search for selective inhibitors. Enzym. Microb. Technol. 2016, 89, 1–6. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Schrödinger Release 2018-4; Schrödinger, LLC.: New York, NY, USA, 2018. [Google Scholar]
- Manta, S.; Xipnitou, A.; Kiritsis, C.; Kantsadi, A.L.; Hayes, J.M.; Skamnaki, V.T.; Lamprakis, C.; Kontou, M.; Zoumpoulakis, P.; Zographos, S.E.; et al. 3′-Axial CH2OH Substitution on Glucopyranose does not Increase Glycogen Phosphorylase Inhibitory Potency. QM/MM-PBSA Calculations Suggest Why. Chem. Biol. Drug Des. 2012, 79, 663–673. [Google Scholar] [CrossRef]
- Murphy, R.B.; Philipp, D.M.; Friesner, R.A. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comput. Chem. 2000, 21, 1442–1457. [Google Scholar] [CrossRef]
- Lin, F.-Y.; MacKerell, J.A.D. Do Halogen–Hydrogen Bond Donor Interactions Dominate the Favorable Contribution of Halogens to Ligand–Protein Binding? J. Phys. Chem. B 2017, 121, 6813–6821. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.; Gu, H.; Wei, N.; Xu, Y.-C.; Fu, W.; Yu, Z. Conformational Preferences of π–π Stacking Between Ligand and Protein, Analysis Derived from Crystal Structure Data Geometric Preference of π–π Interaction. Interdiscip. Sci. Comput. Life Sci. 2015, 7, 211–220. [Google Scholar] [CrossRef]
- Gyöngyösi, T.; Timári, I.; Sinnaeve, D.; Luy, B.; Kövér, K.E. Expedited Nuclear Magnetic Resonance Assignment of Small- to Medium-Sized Molecules with Improved HSQC−CLIP−COSY Experiments. Anal. Chem. 2021, 93, 3096–3102. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors (Short Communication). Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Francl, M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 I |  II | ||||
| Ar | Ki (nM) | Ar | Ki (nM) | ||
| OGA | HexB | OGA | HexA/B a | ||
| Ph (PUGNAc) | 46 [10] | 36 [10] | Ph | 190 [12] | 205 [12] |
| 4-Me-C6H4- | 28 [9] | 21 [9] | 4-Me-C6H4- | 155 [12] | 332 [12] |
| 4-Br-C6H4- | 56 [9] | 47 [9] | 4-Cl-C6H4- | 83 [12] | 170 [12] |
| 2-Naphthyl | 36 [12] | 47 [12] | |||
| Target compounds |  | ||||
 | |||||
| Reagents and conditions: (a) ArSO2NHNH2 (2, 1.5 equiv.), p-TsOH·H2O (0.1 equiv.), CHCl3, reflux; (b) activated MnO2, abs. CH2Cl2, reflux; (c) NH3/MeOH, r.t. | |||||
| Ar | Products and yields (%) | ||||
| 3 | 4 | 5 | Overall yield of 5 from d-glucosamine | ||
| a | p-MePh | 77 | 69 | 94 | 41 |
| b | p-CF3Ph | 85 | 60 | 83 | 35 |
| c | p-FPh | 86 | 72 | 85 | 44 |
| d | p-ClPh | 89 | 77 | 78 | 44 |
| e | 2-naphthyl | 76 | 56 | 75 | 26 |
| f | 1-naphthyl | 89 | 66 | 80 | 39 |
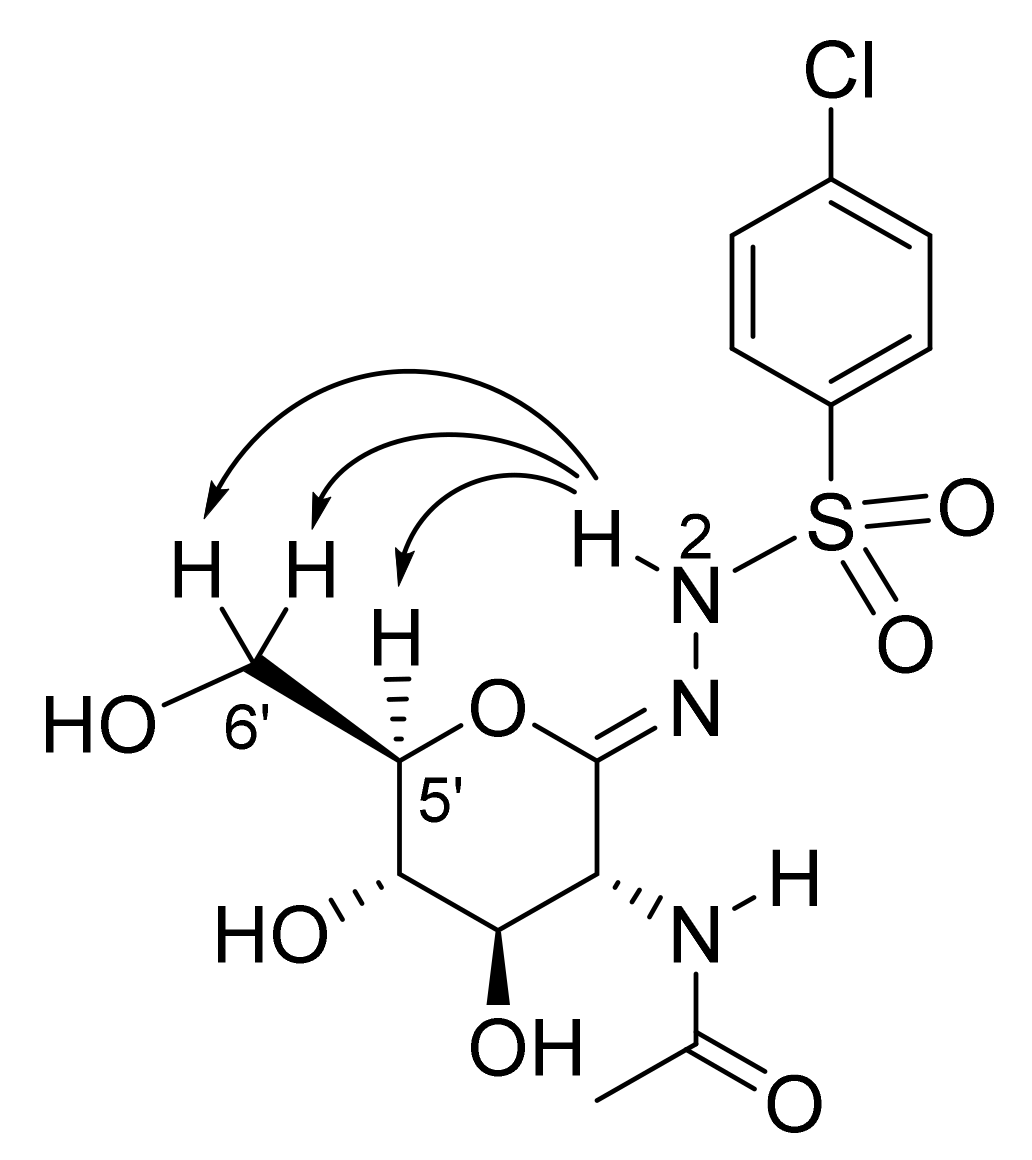
| Compound | Ki (nM) | Ki(hHexB)/ Ki(hOGA)a | ||
| hOGA | hHexB | |||
| PUGNAc |  | 46 [10] | 36 [10] | 0.8 |
| 2-acetamido-2-deoxy- d-glucono-1,5-lactone 4-(2-naphthyl)-semicarbazone |  | 36 [12] | 47 [12] | 1.3 |
| 5a |  | 78 ± 1 | 21 ± 2 | 0.27 |
| 5b |  | 230 ± 17 | 48 ± 4 | 0.21 |
| 5c |  | 95 ± 11 | 45 ± 3 | 0.43 |
| 5d |  | 70 ± 3 | 39 ± 2 | 0.56 |
| 5e |  | 30 ± 2 | 30 ± 3 | 1 |
| 5f |  | 27 ± 7 | 6.8 ± 1.8 | 0.25 |
 | |||||||
| Inhibitor/pose | Prime Hierarchical | Prime Local | QM/MM | ||||
| Prime Energy (kcal/mol) | Relative Energy (kcal/mol) | Prime Energy (kcal/mol) | Relative Energy (kcal/mol) | Absolute Energy (Hartrees) | Relative Energy (kcal/mol) | Dihedral a (°) C1′=N1-N2-H | |
| PUGNAc | |||||||
| Pose 1 | −34,316.1 | 0.0 | −34,320.1 | 0.6 | −1328.97031 | 1.2 | - |
| Pose 2 | −34,315.9 | 0.2 | −34,320.7 | 0.0 | −1328.96987 | 1.5 | - |
| Pose 3 | −34,315.8 | 0.3 | −34,320.1 | 0.6 | −1328.97033 | 1.2 | - |
| Pose 4 | −34,315.6 | 0.5 | −34,319.9 | 0.8 | −1328.97226 | 0.0 | - |
| Pose 5 | −34,315.6 | 0.5 | −34,320.5 | 0.2 | −1328.97227 | 0.0 | - |
| 5a | |||||||
| Pose 1 | −34,317.1 | 0.0 | −34,320.7 | 0.9 | −1728.25188 | 8.8 | −14.9 |
| Pose 2 | −34,315.8 | 1.3 | −34,318.0 | 3.6 | −1728.26592 | 0.0 | −172.2 |
| Pose 3 | −34,315.2 | 1.9 | −34,320.2 | 1.4 | −1728.25192 | 8.8 | −15.1 |
| Pose 4 | −34,315.1 | 2.0 | −34,318.6 | 3.0 | −1728.26597 | 0.0 | −171.8 |
| Pose 5 | −34,314.8 | 2.3 | −34,321.6 | 0.0 | −1728.25203 | 8.7 | −15.1 |
| 5b | |||||||
| Pose 1 | −34,303.5 | 0.0 | −34,310.8 | 0.0 | −2025.91547 | 13.3 | 8.1 |
| Pose 2 | −34,302.4 | 1.1 | −34,304.6 | 6.2 | −2025.93662 | 0.0 | −168.0 |
| Pose 3 | −34,302.0 | 1.5 | −34,305.1 | 5.7 | −2025.92286 | 8.6 | −22.0 |
| Pose 4 | −34,301.5 | 2.0 | −34,305.0 | 5.8 | −2025.92249 | 8.9 | −21.4 |
| Pose 5 | −34,301.5 | 2.0 | −34,305.2 | 5.6 | −2025.92290 | 8.6 | −22.0 |
| 5c | |||||||
| Pose 1 | −34,318.0 | 0.0 | −34,322.8 | 0.3 | −1788.16899 | 8.7 | −17.9 |
| Pose 2 | −34,317.5 | 0.5 | −34,323.1 | 0.0 | −1788.16894 | 8.8 | −17.5 |
| Pose 3 | −34,317.4 | 0.6 | −34,322.3 | 0.8 | −1788.18292 | 0.0 | −170.7 |
| Pose 4 | −34,317.3 | 0.7 | −34,321.8 | 1.3 | −1788.18290 | 0.0 | −170.6 |
| Pose 5 | −34,316.8 | 1.2 | −34,319.1 | 4.0 | −1788.18236 | 0.4 | −149.3 |
| 5d | |||||||
| Pose 1 | −34,318.3 | 0.0 | −34,320.1 | 0.2 | −2148.53075 | 9.0 | −19.6 |
| Pose 2 | −34,316.6 | 1.7 | −34,320.3 | 0.0 | −2148.53084 | 8.9 | −19.7 |
| Pose 3 | −34,315.9 | 2.4 | −34,319.4 | 0.9 | −2148.53121 | 8.7 | −20.5 |
| Pose 4 | −34,315.8 | 2.5 | −34,319.1 | 1.2 | −2148.53073 | 9.0 | −19.3 |
| Pose 5 | −34,315.1 | 3.2 | −34,319.5 | 0.8 | −2148.54505 | 0.0 | −169.2 |
| 5e | |||||||
| Pose 1 | −34,303.0 | 0.0 | −34,308.7 | 0.0 | −1842.54825 | 8.6 | −11.8 |
| Pose 2 | −34,302.9 | 0.1 | −34,306.0 | 2.7 | −1842.56165 | 0.2 | −169.3 |
| Pose 3 | −34,300.8 | 2.2 | −34,306.6 | 2.1 | −1842.56197 | 0.0 | −169.6 |
| Pose 4 | −34,300.6 | 2.4 | −34,306.9 | 1.8 | −1842.56199 | 0.0 | −169.5 |
| Pose 5 | −34,300.3 | 2.7 | −34,306.7 | 2.0 | −1842.56189 | 0.1 | −169.7 |
| 5f | |||||||
| Pose 1 | −34,308.4 | 0.0 | −34,311.4 | 1.5 | −1842.54544 | 4.7 | −0.8 |
| Pose 2 | −34,304.6 | 3.8 | −34,309.9 | 3.0 | −1842.54384 | 5.7 | 3.6 |
| Pose 3 | −34,303.2 | 5.2 | −34,312.9 | 0.0 | −1842.53822 | 9.2 | −2.1 |
| Pose 4 | −34,302.0 | 6.4 | −34,305.5 | 7.4 | −1842.54411 | 5.6 | −169.9 |
| Pose 5 | −34,301.4 | 7.0 | −34,308.7 | 4.2 | −1842.55296 | 0.0 | −154.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiss, M.; Timári, I.; Barna, T.; Mészáros, Z.; Slámová, K.; Bojarová, P.; Křen, V.; Hayes, J.M.; Somsák, L. 2-Acetamido-2-deoxy-d-glucono-1,5-lactone Sulfonylhydrazones: Synthesis and Evaluation as Inhibitors of Human OGA and HexB Enzymes. Int. J. Mol. Sci. 2022, 23, 1037. https://doi.org/10.3390/ijms23031037
Kiss M, Timári I, Barna T, Mészáros Z, Slámová K, Bojarová P, Křen V, Hayes JM, Somsák L. 2-Acetamido-2-deoxy-d-glucono-1,5-lactone Sulfonylhydrazones: Synthesis and Evaluation as Inhibitors of Human OGA and HexB Enzymes. International Journal of Molecular Sciences. 2022; 23(3):1037. https://doi.org/10.3390/ijms23031037
Chicago/Turabian StyleKiss, Mariann, István Timári, Teréz Barna, Zuzana Mészáros, Kristýna Slámová, Pavla Bojarová, Vladimír Křen, Joseph M. Hayes, and László Somsák. 2022. "2-Acetamido-2-deoxy-d-glucono-1,5-lactone Sulfonylhydrazones: Synthesis and Evaluation as Inhibitors of Human OGA and HexB Enzymes" International Journal of Molecular Sciences 23, no. 3: 1037. https://doi.org/10.3390/ijms23031037