
Development in the Mammalian Auditory System Depends on Transcription Factors


 ,
,
Abstract
:1. Introduction

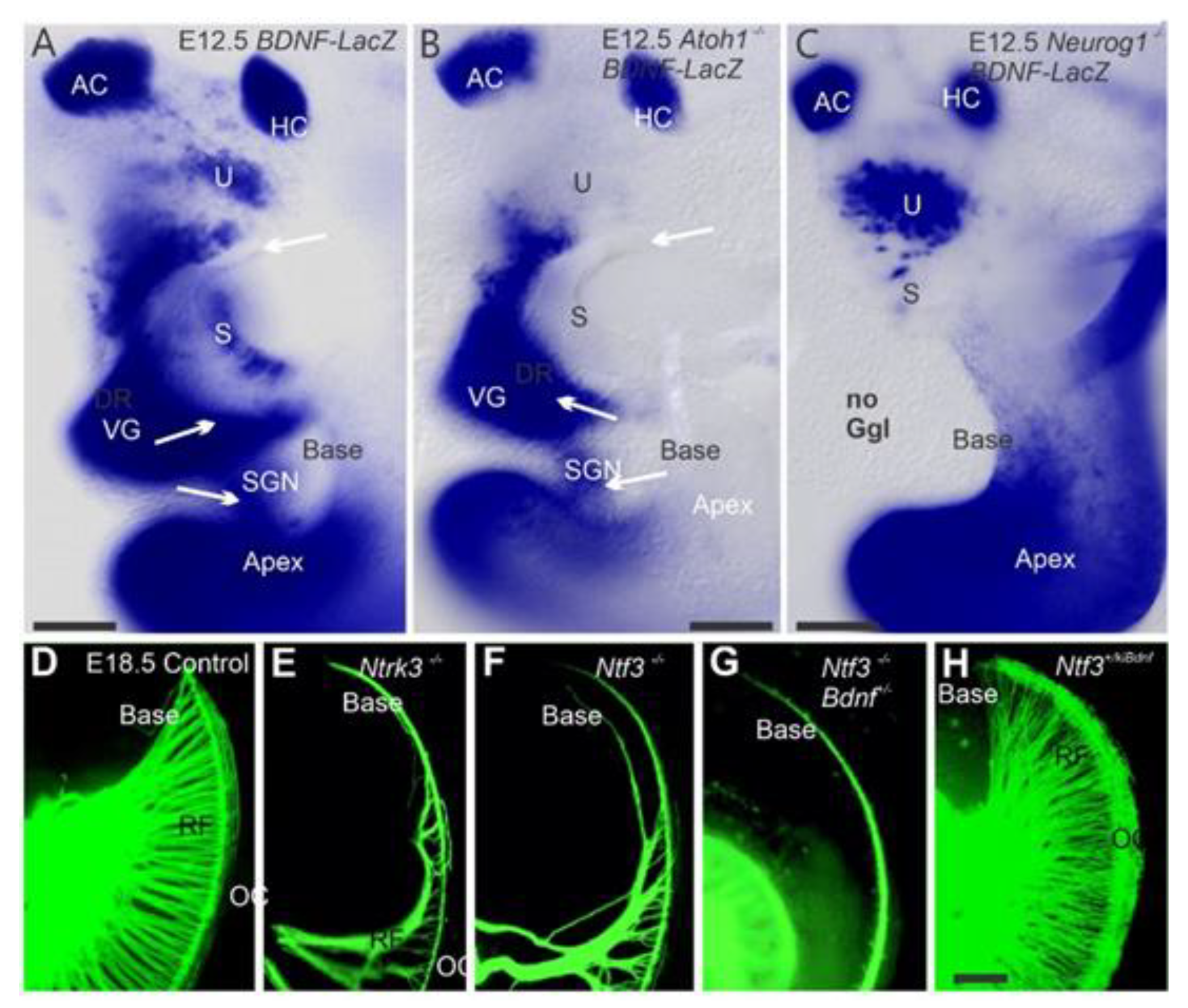
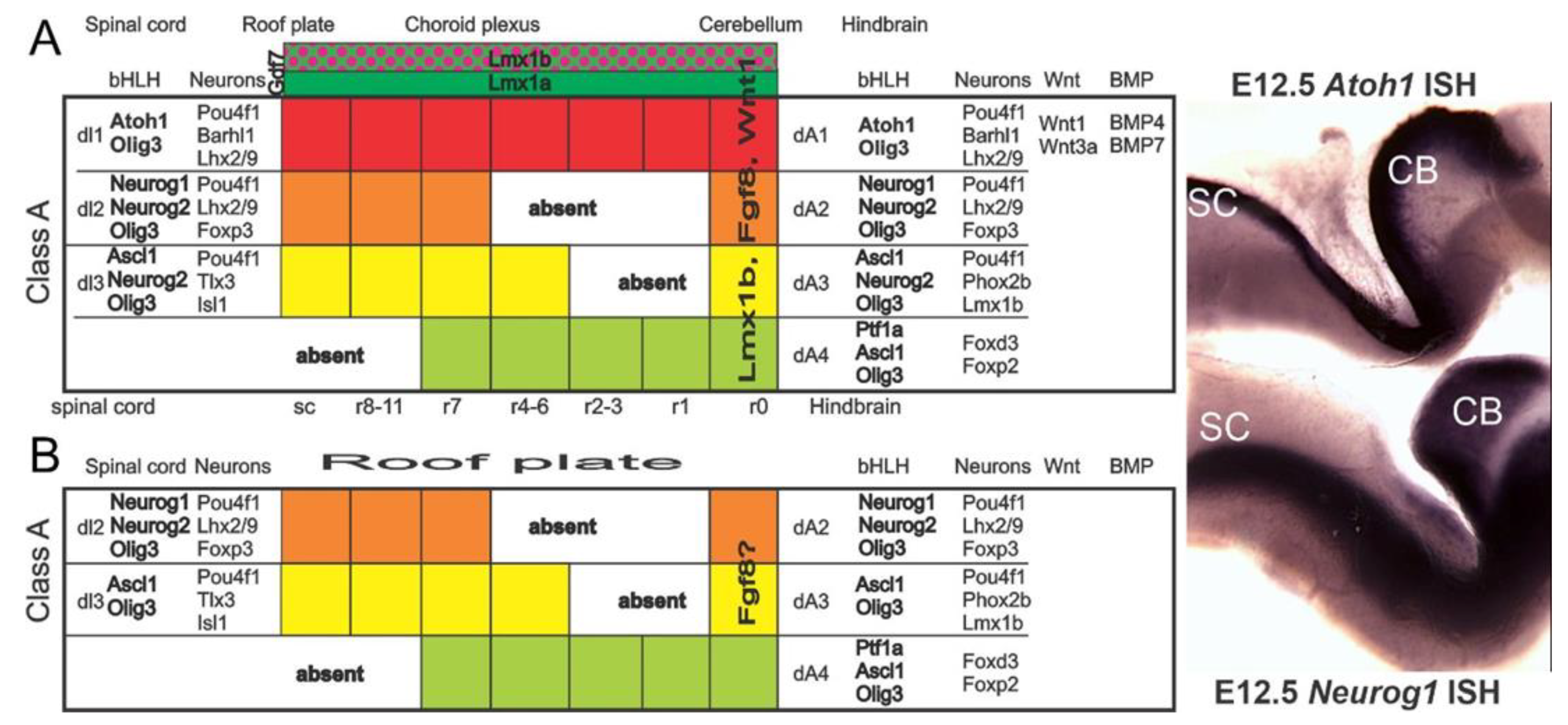
2. Neurog1 Regulates Neurod1 and Atoh1 Expression and Is Essential for Spiral Ganglion Neurons
2.1. Is Atoh1 Playing a Role in Spiral Ganglion Neurons
2.2. Spiral Ganglion Neurons Depend Upon Eya1 and Sox2 and Other Genes
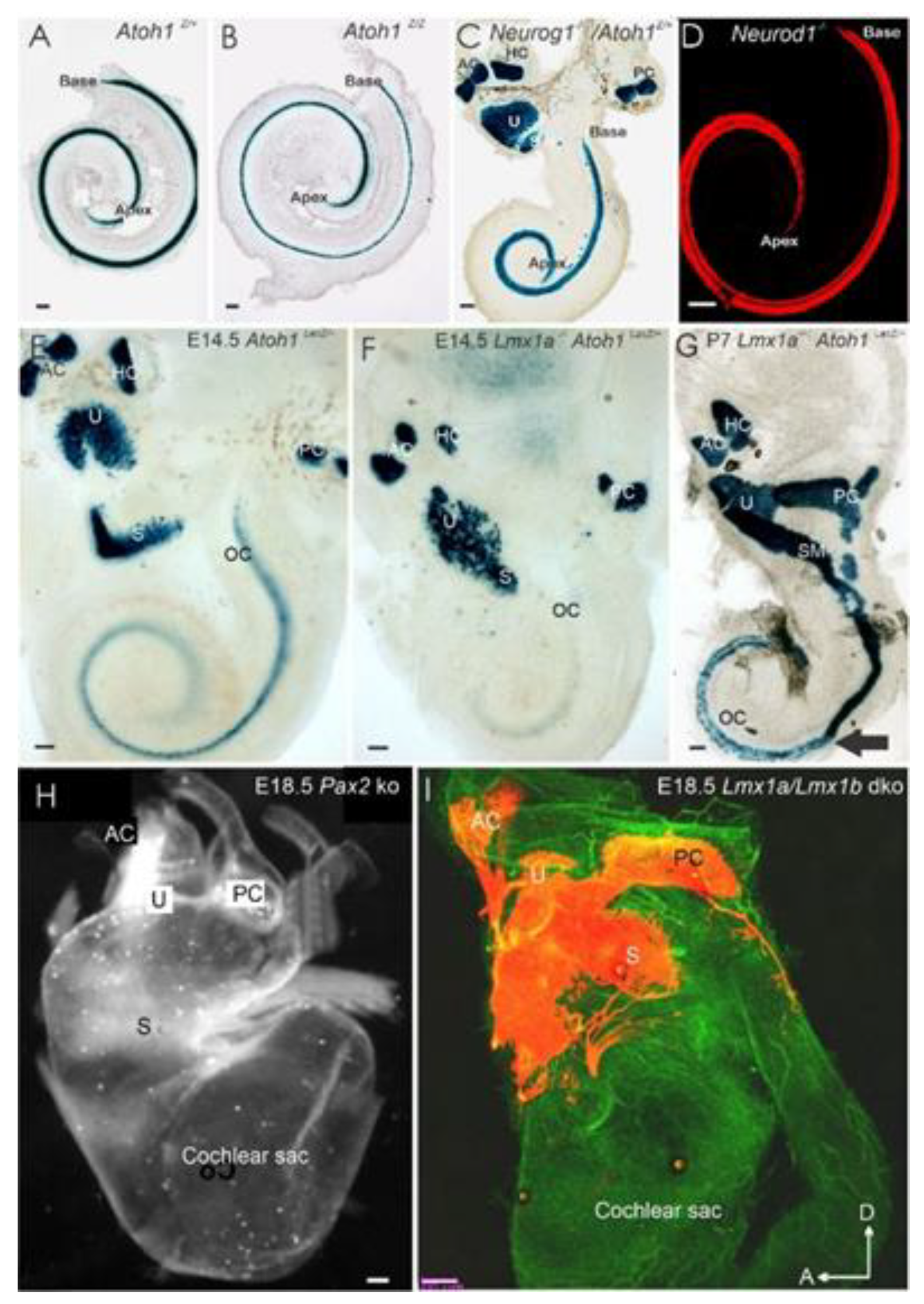
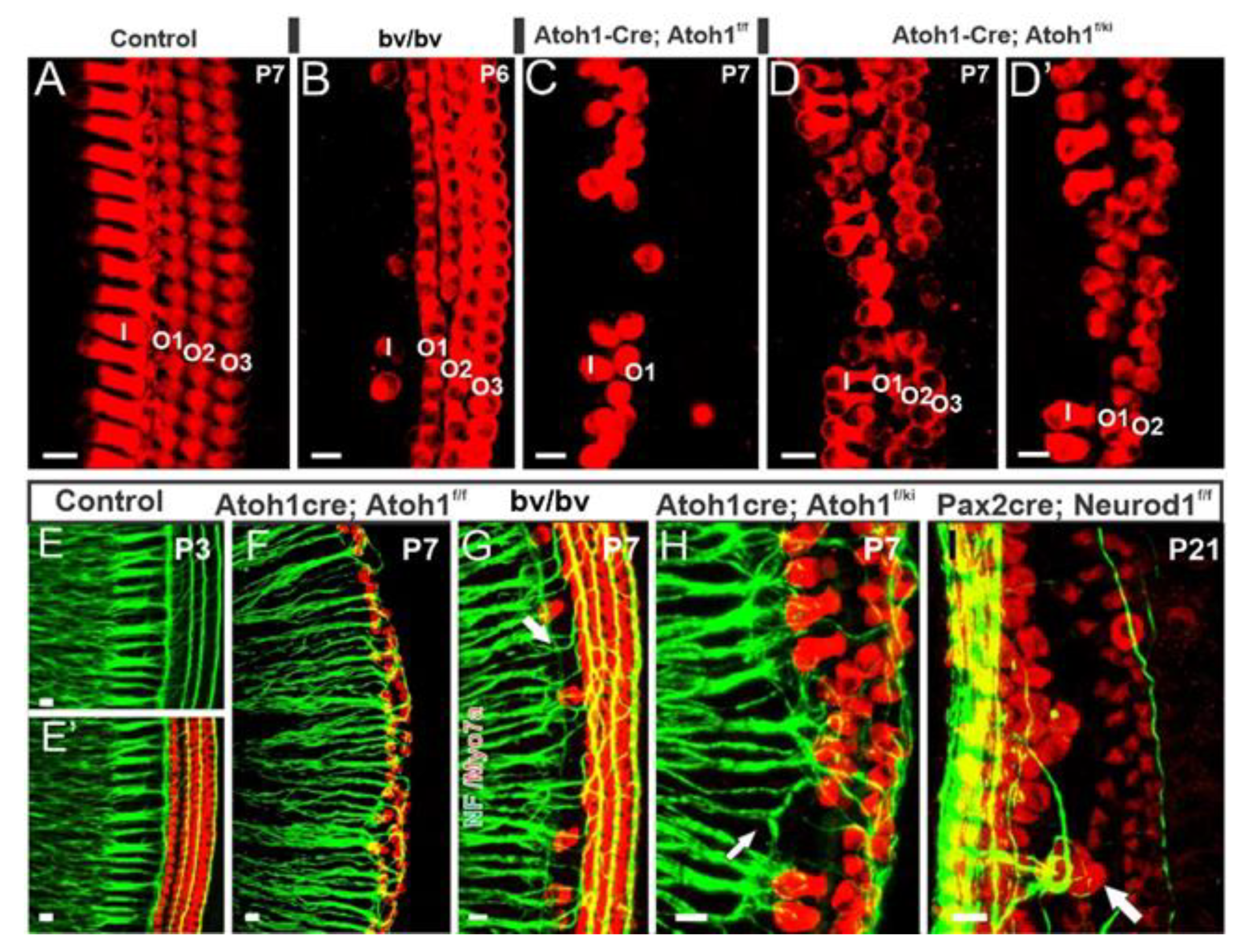
3. Cochlear Hair Cells Require Atoh1
3.1. Neurog1 and Neurod1 Regulate Atoh1 in Cochlear Hair Cells
3.2. Eya1 and Sox2 and Other Transcription Factors Are Essential for HC Development
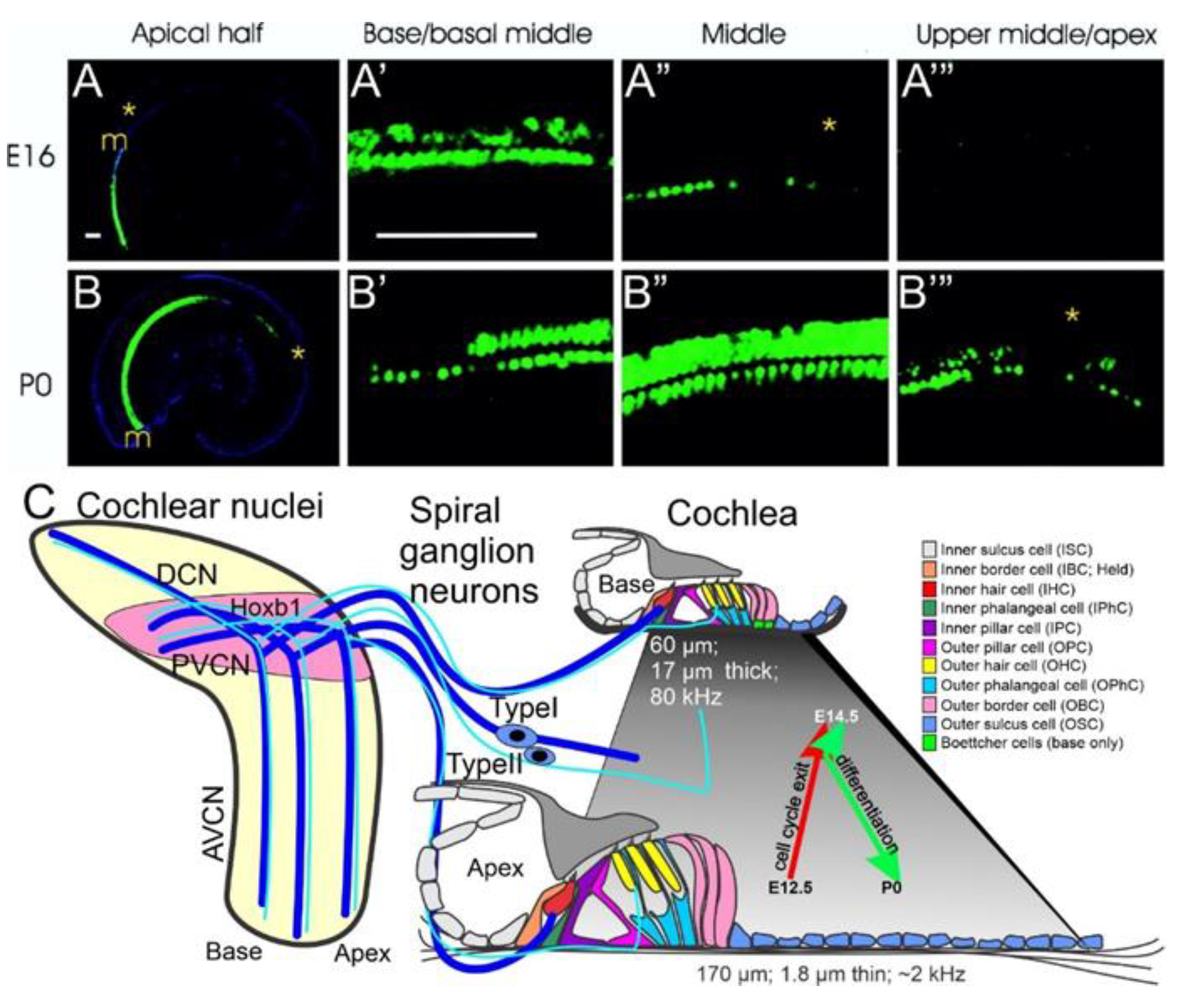
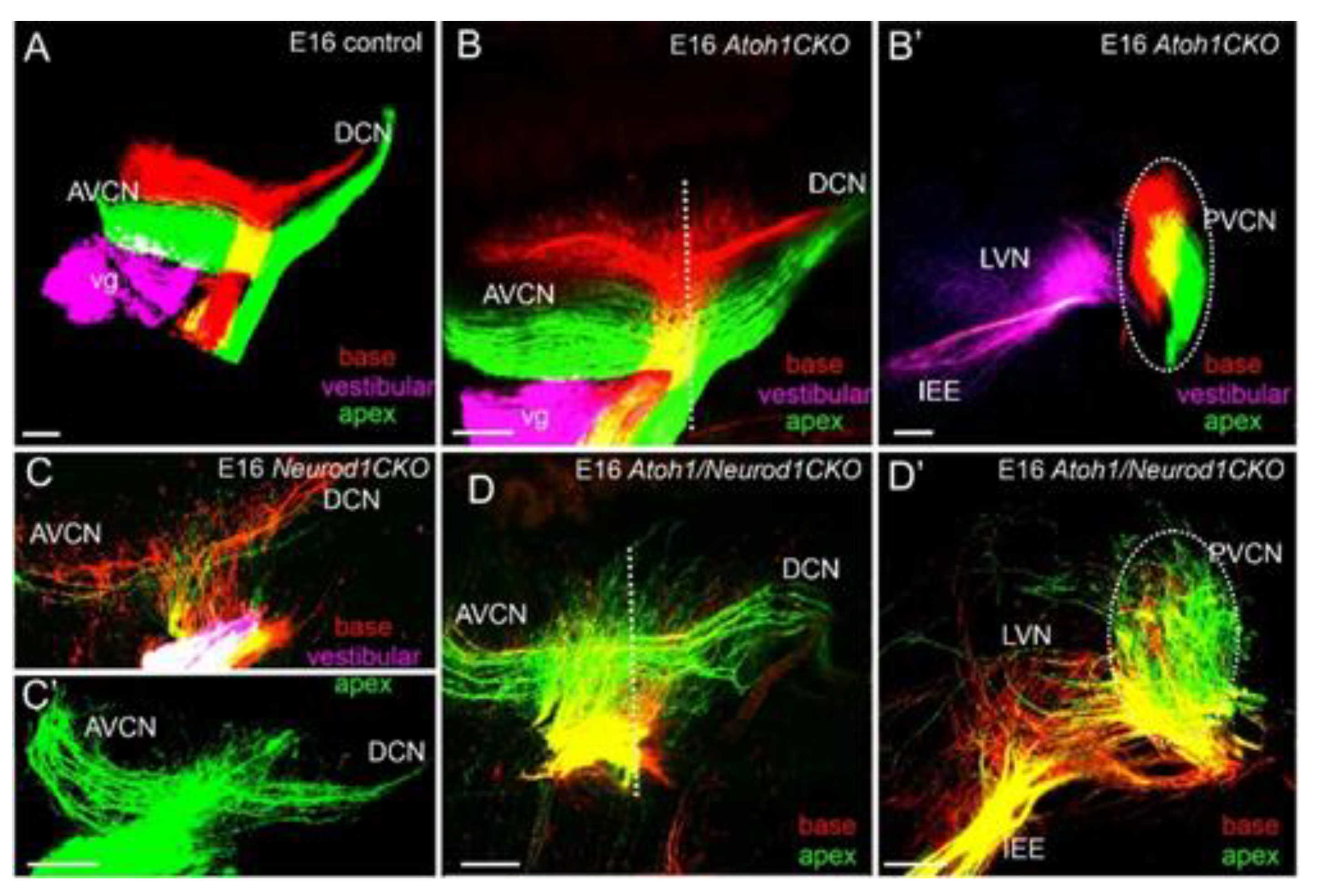
4. Cochlear Nuclei Depend on Atoh1
4.1. Neurod1 Interacts with Atoh1 in the Cochlea
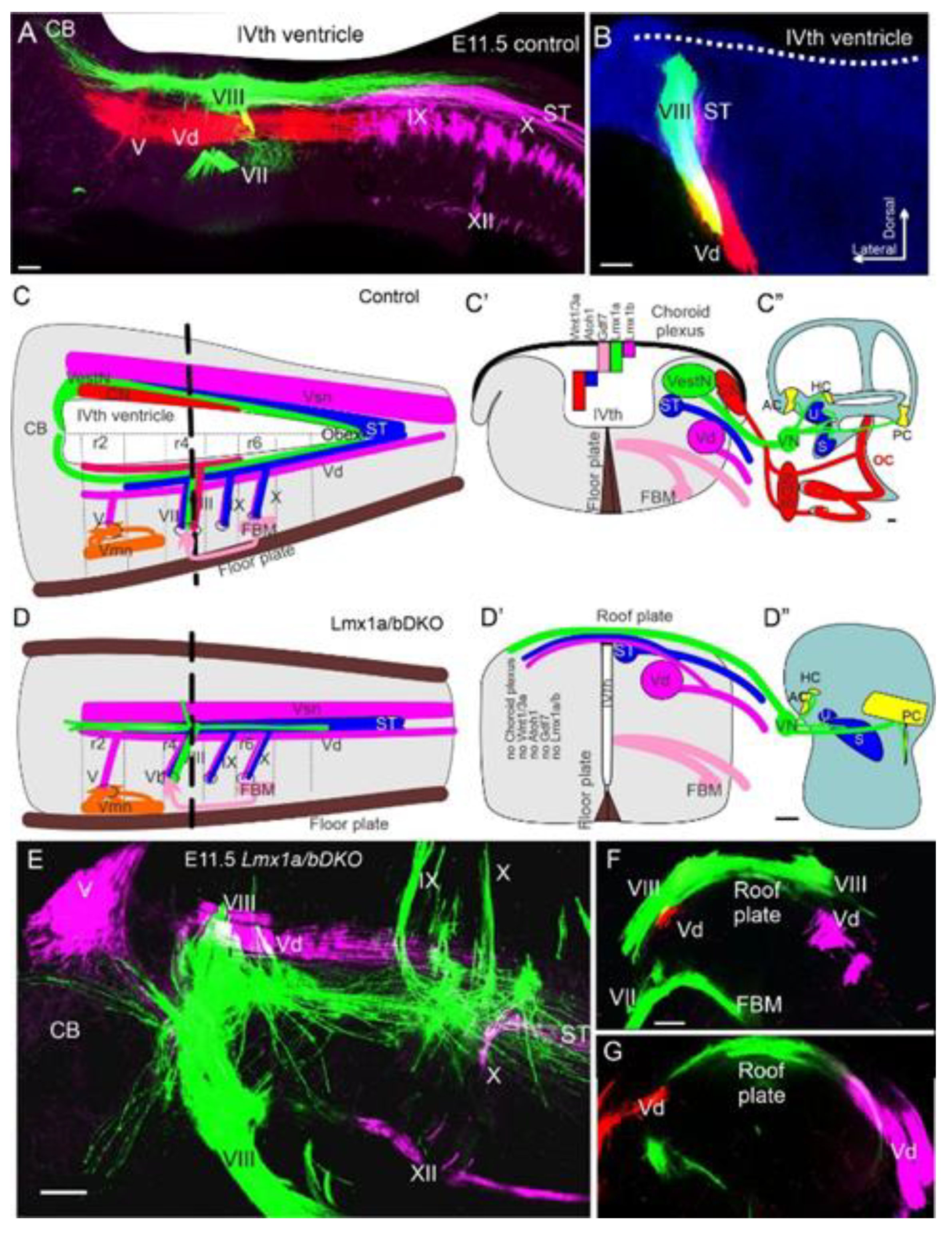
4.2. Sox2 Function Is Unclear, but Lmx1a/b Double Null Mice Eliminate All Cochlear Nuclei
5. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ascl1 | Achete-scute family bHLH transcription factor 1 |
| Atoh1 | Atonal bHLH transcription factor 1 |
| Barhl1 | BarH-like homeobox 1 |
| Bdnf | Brain-derived neurotrophic factor |
| bHLHe22 (bHLHb5) | Basic helix–loop–helix family member e22 |
| Chrna9 | Cholinergic receptor, nicotinic, alpha polypeptide 9 (α9-acetylcholine) |
| Cdkn1b (p27Kip) | Cyclin-dependent kinase inhibitor 1b |
| Dicer1 | Dicer 1, ribonuclease type III |
| Eya1 | EYA transcriptional coactivator, and phosphatase 1 (eyes absent homolog 1) |
| Gata3 | GATA-binding protein 3 |
| Gdf7 | Growth differentiation factor 7 |
| Gfi1 | Growth factor independent 1 transcription repressor |
| Fgf8 | Fibroblast growth factor 8 |
| Fgfr1 | Fibroblast growth factor receptor 1 |
| Foxd3 | Forkhead box D3 |
| Foxg1 | Forkhead box G1 |
| Foxp2 | Forkhead box P2 |
| Fzd3/6 | Frizzled class receptor 3/6 |
| Insm1 | Insulinoma-associated 1 |
| Isl1 | ISL1 transcription factor, LIM/homeodomain |
| Lhx2/9 | LIM homeobox protein 2/9 |
| Lmx1a/b | LIM homeobox transcription factor 1 alpha/beta |
| Myo7a | Myosin VIIA |
| Neurod1 | Neurogenic differentiation 1 |
| Neurog1/2 | Neurogenin 1/2 |
| Nhlh1 | Nescient helix loop helix 1 (Nscl1) |
| Nhlh2 | Nescient helix loop helix 2 (Nscl2) |
| Nmyc2 | Neuroblastoma myc-related oncogene 2 |
| Npr2 | Natriuretic peptide receptor 2 |
| Ntf3 | Neurotrophin 3 (Nt3) |
| Ntrk2 | Neurotrophic tyrosine kinase, receptor, type 2 (TrkB) |
| Ntrk3 | Neurotrophic tyrosine kinase, receptor, type 3 (TrkC) |
| Olig3 | Oligodendrocyte transcription factor 3 |
| Pax2/8 | Paired box 2/8 |
| Phox2b | Paired-like homeobox 2b |
| Pou4f1,3 | POU domain, class 4 transcription factor 1/3 |
| Prickle1 | Prickle planar cell polarity protein 1 |
| Ptf1a | Pancreas-specific transcription factor, 1a |
| Rest | RE1-silencing transcription factor (REST) |
| Six1 | Sine oculis-related homeobox 1 |
| Smarca4 | SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily a, member 4 (Brg1) |
| Sox2 | SRY (sex-determining region Y)-box 2 |
| Srrm4 | Serine/arginine repetitive matrix 4 |
| Tbr1 | T-box brain transcription factor 1 |
| Tlx3 | T cell leukemia, homeobox 3 |
| Vangl2 | VANGL planar cell polarity 2 |
| Wnt1/3a | Wingless-type MMTV integration site family, member 1/3a |
References
- Fritzsch, B.; Pan, N.; Jahan, I.; Duncan, J.S.; Kopecky, B.J.; Elliott, K.L.; Kersigo, J.; Yang, T. Evolution and development of the tetrapod auditory system: An organ of Corti-centric perspective. Evol. Dev. 2013, 15, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.-X.; Manley, G.A. Origins and early evolution of mammalian ears and hearing function. In The Senses; Fritzsch, B., Ed.; Elsevier: Iowa City, NA, USA, 2021; Volume 2, pp. 207–252. [Google Scholar]
- Grothe, B.; Carr, C.E.; Casseday, J.H.; Fritzsch, B.; Köppl, C. The Evolution of Central Pathways and Their Neural Processing Patterns. In Evolution of the Vertebrate Auditory System; Springer Science & Business Media: Berlin, Germany, 2004; pp. 289–359. [Google Scholar]
- Grothe, B. The Auditory System Function—An Integrative Perspective. In The Senses; Fritzsch, B., Ed.; Elsevier: Iowa City, NA, USA, 2021; Volume 2, pp. 1–17. [Google Scholar]
- Zine, A.; Messat, Y.; Fritzsch, B. A human induced pluripotent stem cell-based modular platform to challenge sensorineural hearing loss. Stem Cells 2021. [Google Scholar] [CrossRef]
- Yamoah, E.N.; Li, M.; Shah, A.; Elliott, K.L.; Cheah, K.; Xu, P.-X.; Phillips, S.; Young, S.M., Jr.; Eberl, D.F.; Fritzsch, B. Using Sox2 to alleviate the hallmarks of age-related hearing loss. Ageing Res. Rev. 2020, 59, 101042. [Google Scholar] [CrossRef]
- Resnik, J.; Polley, D.B. Cochlear neural degeneration disrupts hearing in background noise by increasing auditory cortex internal noise. Neuron 2021, 109, 984–996.e4. [Google Scholar] [CrossRef]
- Kersigo, J.; Fritzsch, B. Inner ear hair cells deteriorate in mice engineered to have no or diminished innervation. Front. Aging Neurosci. 2015, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Rubel, E.W.; Fritzsch, B. Auditory system development: Primary auditory neurons and their targets. Annu. Rev. Neurosci. 2002, 25, 51–101. [Google Scholar] [CrossRef] [Green Version]
- De No, R.L. The Primary Acoustic Nuclei; Raven Press: New York, NY, USA, 1981. [Google Scholar]
- Ruben, R.J. Development of the inner ear of the mouse: A radioautographic study of terminal mitoses. Acta Otolaryngol. 1967, 220, 1–44. [Google Scholar]
- Matei, V.; Pauley, S.; Kaing, S.; Rowitch, D.; Beisel, K.; Morris, K.; Feng, F.; Jones, K.; Lee, J.; Fritzsch, B. Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 2005, 234, 633–650. [Google Scholar] [CrossRef] [Green Version]
- Pierce, E.T. Histogenesis of the dorsal and ventral cochlear nuclei in the mouse. An autoradiographic study. J. Comp. Neurol. 1967, 131, 27–53. [Google Scholar] [CrossRef]
- Fritzsch, B.; Elliott, K.L.; Pavlinkova, G. Primary sensory map formations reflect unique needs and molecular cues specific to each sensory system. F1000Reseach 2019, 8. [Google Scholar] [CrossRef]
- Ma, Q.; Anderson, D.J.; Fritzsch, B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J. Assoc. Res. Otolaryngol. 2000, 1, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-Y.; Fritzsch, B.; Serls, A.; Bakel, L.A.; Huang, E.J.; Reichardt, L.F.; Barth, D.S.; Lee, J.E. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development 2001, 128, 417–426. [Google Scholar]
- Ma, Q.; Chen, Z.; del Barco Barrantes, I.; de la Pompa, J.L.; Anderson, D.J. neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 1998, 20, 469–482. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Pereira, F.A.; Price, S.D.; Chu, M.-j.; Shope, C.; Himes, D.; Eatock, R.A.; Brownell, W.E.; Lysakowski, A.; Tsai, M.-J. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000, 14, 2839–2854. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Treadaway, J.; Buckner, T.W.; Fritzsch, B. Visualization of α9 acetylcholine receptor expression in hair cells of transgenic mice containing a modified bacterial artificial chromosome. Proc. Natl. Acad. Sci.USA 1999, 96, 14100–14105. [Google Scholar] [CrossRef] [Green Version]
- Booth, K.T.; Azaiez, H.; Jahan, I.; Smith, R.J.; Fritzsch, B. Intracellular Regulome Variability Along the Organ of Corti: Evidence, Approaches, Challenges, and Perspective. Front. Genet. 2018, 9, 156. [Google Scholar] [CrossRef]
- Muniak, M.A.; Connelly, C.J.; Suthakar, K.; Milinkeviciute, G.; Ayeni, F.E.; Ryugo, D.K. Central Projections of Spiral Ganglion Neurons. In The Primary Auditory Neurons of the Mammalian Cochlea; Springer: New York, NY, USA, 2016; pp. 157–190. [Google Scholar]
- Lowenheim, H.; Furness, D.N.; Kil, J.; Zinn, C.; Gultig, K.; Fero, M.L.; Frost, D.; Gummer, A.W.; Roberts, J.M.; Rubel, E.W.; et al. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of corti. Proc. Natl. Acad. Sci.USA 1999, 96, 4084–4088. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Segil, N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development 1999, 126, 1581–1590. [Google Scholar]
- Bermingham, N.A.; Hassan, B.A.; Price, S.D.; Vollrath, M.A.; Ben-Arie, N.; Eatock, R.A.; Bellen, H.J.; Lysakowski, A.; Zoghbi, H.Y. Math1: An essential gene for the generation of inner ear hair cells. Science 1999, 284, 1837–1841. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Johnson, J.E.; Zoghbi, H.Y.; Segil, N. The role of Math1 in inner ear development: Uncoupling the establishment of the sensory primordium from hair cell fate determination. Development 2002, 129, 2495–2505. [Google Scholar]
- Fritzsch, B.; Beisel, K.W.; Bermingham, N.A. Developmental evolutionary biology of the vertebrate ear: Conserving mechanoelectric transduction and developmental pathways in diverging morphologies. Neuroreport 2000, 11, R35–R44. [Google Scholar] [CrossRef] [PubMed]
- Fritzsch, B.; Beisel, K.W.; Hansen, L.A. The molecular basis of neurosensory cell formation in ear development: A blueprint for hair cell and sensory neuron regeneration? Bioessays 2006, 28, 1181–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahan, I.; Pan, N.; Kersigo, J.; Fritzsch, B. Neurod1 suppresses hair cell differentiation in ear ganglia and regulates hair cell subtype development in the cochlea. PLoS ONE 2010, 5, e11661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzsch, B.; Eberl, D.F.; Beisel, K.W. The role of bHLH genes in ear development and evolution: Revisiting a 10-year-old hypothesis. Cell. Mol. Life Sci. CMLS 2010, 67, 3089–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
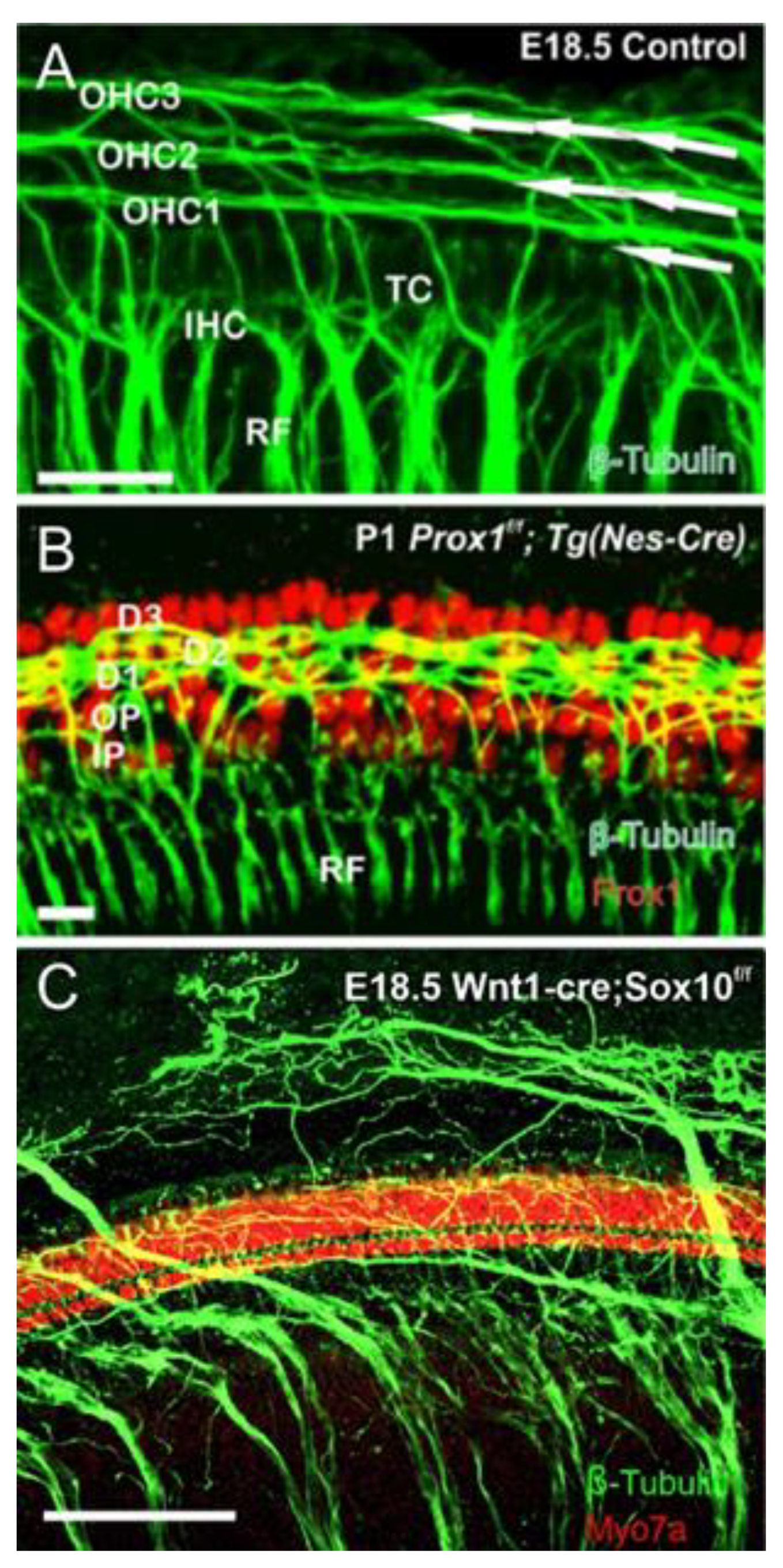
- Fritzsch, B.; Dillard, M.; Lavado, A.; Harvey, N.L.; Jahan, I. Canal cristae growth and fiber extension to the outer hair cells of the mouse ear require Prox1 activity. PLoS ONE 2010, 5, e9377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecky, B.J.; Jahan, I.; Fritzsch, B. Correct timing of proliferation and differentiation is necessary for normal inner ear development and auditory hair cell viability. Dev. Dyn. 2013, 242, 132–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecky, B.; Santi, P.; Johnson, S.; Schmitz, H.; Fritzsch, B. Conditional deletion of N-Myc disrupts neurosensory and non-sensory development of the ear. Dev. Dyn. 2011, 240, 1373–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, V.Y.; Rose, M.F.; Zoghbi, H.Y. Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron 2005, 48, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Karis, A.; Pata, I.; van Doorninck, J.H.; Grosveld, F.; de Zeeuw, C.I.; de Caprona, D.; Fritzsch, B. Transcription factor GATA-3 alters pathway selection of olivocochlear neurons and affects morphogenesis of the ear. J. Comp. Neurol. 2001, 429, 615–630. [Google Scholar] [CrossRef]
- Maklad, A.; Fritzsch, B. Development of vestibular afferent projections into the hindbrain and their central targets. Brain Res. Bull. 2003, 60, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Jahan, I.; Kersigo, J.; Pan, N.; Fritzsch, B. Neurod1 regulates survival and formation of connections in mouse ear and brain. Cell Tissue Res. 2010, 341, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Fritzsch, B.; Pauley, S.; Feng, F.; Matei, V.; Nichols, D. The molecular and developmental basis of the evolution of the vertebrate auditory system. Int. J. Comp. Psychol. 2006, 19, 1–25. [Google Scholar]
- Pan, N.; Jahan, I.; Lee, J.E.; Fritzsch, B. Defects in the cerebella of conditional Neurod1 null mice correlate with effective Tg (Atoh1-cre) recombination and granule cell requirements for Neurod1 for differentiation. Cell Tissue Res. 2009, 337, 407–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiyama, T.; Yamada, M.; Terao, M.; Terashima, T.; Hioki, H.; Inoue, Y.U.; Inoue, T.; Masuyama, N.; Obata, K.; Yanagawa, Y. Inhibitory and excitatory subtypes of cochlear nucleus neurons are defined by distinct bHLH transcription factors, Ptf1a and Atoh1. Development 2009, 136, 2049–2058. [Google Scholar] [CrossRef] [Green Version]
- Mishima, Y.; Lindgren, A.G.; Chizhikov, V.V.; Johnson, R.L.; Millen, K.J. Overlapping function of Lmx1a and Lmx1b in anterior hindbrain roof plate formation and cerebellar growth. J. Neurosci. 2009, 29, 11377–11384. [Google Scholar] [CrossRef] [Green Version]
- Koehler, K.R.; Nie, J.; Longworth-Mills, E.; Liu, X.-P.; Lee, J.; Holt, J.R.; Hashino, E. Generation of inner ear organoids containing functional hair cells from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 583. [Google Scholar] [CrossRef] [Green Version]
- Macova, I.; Pysanenko, K.; Chumak, T.; Dvorakova, M.; Bohuslavova, R.; Syka, J.; Fritzsch, B.; Pavlinkova, G. Neurod1 is essential for the primary tonotopic organization and related auditory information processing in the midbrain. J. Neurosci. 2019, 39, 984–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filova, I.; Dvorakova, M.; Bohuslavova, R.; Pavlinek, A.; Elliott, K.L.; Vochyanova, S.; Fritzsch, B.; Pavlinkova, G. Combined Atoh1 and Neurod1 Deletion Reveals Autonomous Growth of Auditory Nerve Fibers. Mol. Neurobiol. 2020, 57, 5307–5323. [Google Scholar] [CrossRef]
- Jahan, I.; Elliott, K.L.; Fritzsch, B. Understanding molecular evolution and development of the organ of Corti can provide clues for hearing restoration. Integr. Comp. Biol. 2018, 58, 351–365. [Google Scholar] [CrossRef]
- Li, H.J.; Ray, S.K.; Pan, P.; Haigh, J.; Fritzsch, B.; Leiter, A.B. Intestinal Neurod1 expression impairs paneth cell differentiation and promotes enteroendocrine lineage specification. Sci. Rep. 2019, 9, 19489. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Kersigo, J.; Jahan, I.; Pan, N.; Fritzsch, B. The molecular basis of making spiral ganglion neurons and connecting them to hair cells of the organ of Corti. Hear. Res. 2011, 278, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Fritzsch, B.; Kersigo, J.; Yang, T.; Jahan, I.; Pan, N. Neurotrophic Factor Function during Ear Development: Expression Changes Define Critical Phases for Neuronal Viability. In The Primary Auditory Neurons of the Mammalian Cochlea; Springer: New York, NY, USA, 2016; pp. 49–84. [Google Scholar]
- Fariñas, I.; Jones, K.R.; Tessarollo, L.; Vigers, A.J.; Huang, E.; Kirstein, M.; De Caprona, D.C.; Coppola, V.; Backus, C.; Reichardt, L.F. Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. J. Neurosci. 2001, 21, 6170–6180. [Google Scholar] [CrossRef] [Green Version]
- Fritzsch, B.; Silos-Santiago, I.; Bianchi, L.M.; Farinas, I. The role of neurotrophic factors in regulating the development of inner ear innervation. Trends Neurosci. 1997, 20, 159–164. [Google Scholar] [CrossRef]
- Silos-Santiago, I.; Fagan, A.M.; Garber, M.; Fritzsch, B.; Barbacid, M. Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur. J. Neurosci. 1997, 9, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Coate, T.M.; Kelley, M.W. Making Connections in the Inner ear: Recent Insights into the Development of Spiral Ganglion Neurons and Their Connectivity with Sensory Hair Cells. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2013; Volume 26, pp. 460–469. [Google Scholar]
- Goodrich, L.V. Early Development of the Spiral Ganglion. In The Primary Auditory Neurons of the Mammalian Cochlea; Springer: New York, NY, USA, 2016; pp. 11–48. [Google Scholar]
- Jung, J.S.; Zhang, K.D.; Wang, Z.; McMurray, M.; Tkaczuk, A.; Ogawa, Y.; Hertzano, R.; Coate, T.M. Semaphorin-5B controls spiral ganglion neuron branch refinement during development. J. Neurosci. 2019, 39, 6425–6438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druckenbrod, N.; Hale, E.; Olukoya, O.; Shatzer, W.; Goodrich, L. Neuronal processes and glial precursors form a scaffold for wiring the developing mouse cochlea. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Rodriguez, E.R.; Reimert, D.V.; Shu, T.; Fritzsch, B.; Richards, L.J.; Kolodkin, A.L.; Ginty, D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 2003, 5, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Noda, T.; Dabdoub, A. Dynamic expression of Sox2, Gata3, and Prox1 during primary auditory neuron development in the mammalian cochlea. PLoS ONE 2017, 12, e0170568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Reiprich, S.; Wegner, M.; Fritzsch, B. Targeted deletion of Sox10 by Wnt1-cre defects neuronal migration and projection in the mouse inner ear. PLoS ONE 2014, 9, e94580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghimire, S.R.; Deans, M.R. Frizzled3 and Frizzled6 Cooperate with Vangl2 to Direct Cochlear Innervation by type II Spiral Ganglion Neurons. J. Neurosci. 2019, 39, 8013–8023. [Google Scholar] [CrossRef] [Green Version]
- Jahan, I.; Pan, N.; Kersigo, J.; Calisto, L.E.; Morris, K.A.; Kopecky, B.; Duncan, J.S.; Beisel, K.W.; Fritzsch, B. Expression of Neurog1 instead of Atoh1 can partially rescue organ of Corti cell survival. PLoS ONE 2012, 7, e30853. [Google Scholar] [CrossRef] [Green Version]
- Jahan, I.; Pan, N.; Kersigo, J.; Fritzsch, B. Neurog1 can partially substitute for Atoh1 function in hair cell differentiation and maintenance during organ of Corti development. Development 2015, 142, 2810–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, M.C. Noise-induced and age-related hearing loss: New perspectives and potential therapies. F1000Reseach 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Jadali, A.; Fritzsch, B.; Kwan, K.Y. NEUROG1 Regulates CDK2 to Promote Proliferation in Otic Progenitors. Stem Cell Rep. 2017, 9, 1516–1529. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ueno, H.; Xu, C.Y.; Chen, B.; Weissman, I.L.; Xu, P.X. Identification of mouse cochlear progenitors that develop hair and supporting cells in the organ of Corti. Nat Commun 2017, 8, 15046. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Maklad, A.; Hansen, L.A.; Feng, F.; Sorensen, C.; Lee, K.-F.; Macklin, W.B.; Fritzsch, B. A disorganized innervation of the inner ear persists in the absence of ErbB2. Brain Res. 2006, 1091, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Nakano, Y.; Jahan, I.; Bonde, G.; Sun, X.; Hildebrand, M.S.; Engelhardt, J.F.; Smith, R.J.; Cornell, R.A.; Fritzsch, B.; Bánfi, B. A mutation in the Srrm4 gene causes alternative splicing defects and deafness in the Bronx waltzer mouse. PLoS Genet 2012, 8, e1002966. [Google Scholar] [CrossRef]
- Bouchard, M.; de Caprona, D.; Busslinger, M.; Xu, P.; Fritzsch, B. Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev. Biol. 2010, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Chizhikov, V.V.; Iskusnykh, I.Y.; Fattakhov, N.; Fritzsch, B. Lmx1a and Lmx1b are Redundantly Required for the Development of Multiple Components of the Mammalian Auditory System. Neuroscience 2021, 452, 247–264. [Google Scholar] [CrossRef]
- Fritzsch, B.; Matei, V.; Nichols, D.; Bermingham, N.; Jones, K.; Beisel, K.; Wang, V. Atoh1 null mice show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2005, 233, 570–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, D.H.; Pauley, S.; Jahan, I.; Beisel, K.W.; Millen, K.J.; Fritzsch, B. Lmx1a is required for segregation of sensory epithelia and normal ear histogenesis and morphogenesis. Cell Tissue Res. 2008, 334, 339–358. [Google Scholar] [CrossRef] [Green Version]
- Xiang, M.; Maklad, A.; Pirvola, U.; Fritzsch, B. Brn3c null mutant mice show long-term, incomplete retention of some afferent inner ear innervation. BMC Neurosci. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Pauley, S.; Kopecky, B.; Beisel, K.; Soukup, G.; Fritzsch, B. Stem cells and molecular strategies to restore hearing. Panminerva Med. 2008, 50, 41. [Google Scholar] [PubMed]
- Pan, N.; Jahan, I.; Kersigo, J.; Kopecky, B.; Santi, P.; Johnson, S.; Schmitz, H.; Fritzsch, B. Conditional deletion of Atoh1 using Pax2-Cre results in viable mice without differentiated cochlear hair cells that have lost most of the organ of Corti. Hear. Res. 2011, 275, 66–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, N.; Jahan, I.; Kersigo, J.; Duncan, J.S.; Kopecky, B.; Fritzsch, B. A novel Atoh1 “self-terminating” mouse model reveals the necessity of proper Atoh1 level and duration for hair cell differentiation and viability. PLoS ONE 2012, 7, e30358. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.B.; Cortez, S.R.; Beyer, L.A.; Wiler, J.A.; Di Polo, A.; Pfingst, B.E.; Raphael, Y. Transgenic BDNF induces nerve fiber regrowth into the auditory epithelium in deaf cochleae. Exp. Neurol. 2010, 223, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Puligilla, C.; Dabdoub, A.; Brenowitz, S.D.; Kelley, M.W. Sox2 induces neuronal formation in the developing mammalian cochlea. J. Neurosci. 2010, 30, 714–722. [Google Scholar] [CrossRef]
- Steevens, A.R.; Sookiasian, D.L.; Glatzer, J.C.; Kiernan, A.E. SOX2 is required for inner ear neurogenesis. Sci. Rep. 2017, 7, 4086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Li, J.; Zhang, T.; Jiang, H.; Ramakrishnan, A.; Fritzsch, B.; Shen, L.; Xu, P.X. Chromatin remodelers and lineage-specific factors interact to target enhancers to establish proneurosensory fate within otic ectoderm. Proc. Natl. Acad. Sci.USA 2021, 118. [Google Scholar] [CrossRef]
- Dvorakova, M.; Macova, I.; Bohuslavova, R.; Anderova, M.; Fritzsch, B.; Pavlinkova, G. Early ear neuronal development, but not olfactory or lens development, can proceed without SOX2. Dev. Biol. 2020, 457, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, M.; Jahan, I.; Macova, I.; Chumak, T.; Bohuslavova, R.; Syka, J.; Fritzsch, B.; Pavlinkova, G. Incomplete and delayed Sox2 deletion defines residual ear neurosensory development and maintenance. Sci. Rep. 2016, 6, 38253. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, A.E.; Pelling, A.L.; Leung, K.K.; Tang, A.S.; Bell, D.M.; Tease, C.; Lovell-Badge, R.; Steel, K.P.; Cheah, K.S. Sox2 is required for sensory organ development in the mammalian inner ear. Nature 2005, 434, 1031. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Silvius, D.; Fritzsch, B.; Xu, P.-X. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development 2004, 131, 5561–5572. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Kersigo, J.; Wu, S.; Fritzsch, B.; Bassuk, A.G. Prickle1 regulates neurite outgrowth of apical spiral ganglion neurons but not hair cell polarity in the murine cochlea. PLoS ONE 2017, 12, e0183773. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.S.; Fritzsch, B. Continued expression of GATA3 is necessary for cochlear neurosensory development. PLoS ONE 2013, 8, e62046. [Google Scholar] [CrossRef] [Green Version]
- Soukup, G.A.; Fritzsch, B.; Pierce, M.L.; Weston, M.D.; Jahan, I.; McManus, M.T.; Harfe, B.D. Residual microRNA expression dictates the extent of inner ear development in conditional Dicer knockout mice. Dev. Biol. 2009, 328, 328–341. [Google Scholar] [CrossRef] [Green Version]
- Kersigo, J.; D’Angelo, A.; Gray, B.D.; Soukup, G.A.; Fritzsch, B. The role of sensory organs and the forebrain for the development of the craniofacial shape as revealed by Foxg1-cre-mediated microRNA loss. Genesis 2011, 49, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-S.; Liu, F.; Segil, N. A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development 2006, 133, 2817–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateya, T.; Sakamoto, S.; Ishidate, F.; Hirashima, T.; Imayoshi, I.; Kageyama, R. Three-dimensional live imaging of Atoh1 reveals the dynamics of hair cell induction and organization in the developing cochlea. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Dabdoub, A.; Puligilla, C.; Jones, J.M.; Fritzsch, B.; Cheah, K.S.; Pevny, L.H.; Kelley, M.W. Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc. Natl. Acad. Sci.USA 2008, 105, 18396–18401. [Google Scholar] [CrossRef] [Green Version]
- Driver, E.C.; Sillers, L.; Coate, T.M.; Rose, M.F.; Kelley, M.W. The Atoh1-lineage gives rise to hair cells and supporting cells within the mammalian cochlea. Dev. Biol. 2013, 376, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driver, E.C.; Northrop, A.; Kelley, M.W. Cell migration, intercalation and growth regulate mammalian cochlear extension. Development 2017, 144, 3766–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, T.; Seymour, M.L.; Zhang, H.; Pereira, F.A.; Groves, A.K. Conditional deletion of Atoh1 reveals distinct critical periods for survival and function of hair cells in the organ of Corti. J. Neurosci. 2013, 33, 10110–10122. [Google Scholar] [CrossRef] [Green Version]
- Chonko, K.T.; Jahan, I.; Stone, J.; Wright, M.C.; Fujiyama, T.; Hoshino, M.; Fritzsch, B.; Maricich, S.M. Atoh1 directs hair cell differentiation and survival in the late embryonic mouse inner ear. Dev. Biol. 2013, 381, 401–410. [Google Scholar] [CrossRef]
- Kelly, M.C.; Chang, Q.; Pan, A.; Lin, X.; Chen, P. Atoh1 directs the formation of sensory mosaics and induces cell proliferation in the postnatal mammalian cochlea in vivo. J. Neurosci. 2012, 32, 6699–6710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.M.; Doetzlhofer, A.; Lee, Y.S.; Groves, A.K.; Segil, N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature 2006, 441, 984. [Google Scholar] [CrossRef]
- Nakano, Y.; Wiechert, S.; Fritzsch, B.; Bánfi, B. Inhibition of a transcriptional repressor rescues hearing in a splicing factor-deficient mouse. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Dabdoub, A.; Nishimura, K. Cochlear implants meet regenerative biology: State of the science and future research directions. Otol. Neurotol. 2017, 38, e232–e236. [Google Scholar] [CrossRef]
- Yamashita, T.; Zheng, F.; Finkelstein, D.; Kellard, Z.; Carter, R.; Rosencrance, C.D.; Sugino, K.; Easton, J.; Gawad, C.; Zuo, J. High-resolution transcriptional dissection of in vivo Atoh1-mediated hair cell conversion in mature cochleae identifies Isl1 as a co-reprogramming factor. PLoS Genet. 2018, 14, e1007552. [Google Scholar] [CrossRef]
- Walters, B.J.; Coak, E.; Dearman, J.; Bailey, G.; Yamashita, T.; Kuo, B.; Zuo, J. In vivo interplay between p27Kip1, GATA3, ATOH1, and POU4F3 converts non-sensory cells to hair cells in adult mice. Cell Rep. 2017, 19, 307–320. [Google Scholar] [CrossRef]
- Lopez-Juarez, A.; Lahlou, H.; Ripoll, C.; Cazals, Y.; Brezun, J.M.; Wang, Q.; Edge, A.; Zine, A. Engraftment of Human Stem Cell-Derived Otic Progenitors in the Damaged Cochlea. Mol. Ther. 2019, 27, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Lenz, D.R.; Gunewardene, N.; Abdul-Aziz, D.E.; Wang, Q.; Gibson, T.M.; Edge, A.S. Applications of Lgr5-positive cochlear progenitors (LCPs) to the study of hair cell differentiation. Front. Cell Dev. Biol. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccio, M.; Senn, P.; Heller, S. Novel insights into inner ear development and regeneration for refined hearing loss therapies. Hear. Res. 2019, 397, 107859. [Google Scholar]
- Zhang, J.; Wang, Q.; Abdul-Aziz, D.; Mattiacio, J.; Edge, A.S.; White, P.M. ERBB 2 signaling drives supporting cell proliferation in vitro and apparent supernumerary hair cell formation in vivo in the neonatal mouse cochlea. Eur. J. Neurosci. 2018, 48, 3299–3316. [Google Scholar] [CrossRef] [PubMed]
- Schilder, A.G.; Su, M.P.; Blackshaw, H.; Lustig, L.; Staecker, H.; Lenarz, T.; Safieddine, S.; Gomes-Santos, C.S.; Holme, R.; Warnecke, A. Hearing Protection, Restoration, and Regeneration: An Overview of Emerging Therapeutics for Inner Ear and Central Hearing Disorders. Otol. Neurotol. 2019, 40, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.A.; Groves, A.K. Transcription Factor Reprogramming in the Inner Ear: Turning on Cell Fate Switches to Regenerate Sensory Hair Cells. Front. Cell. Neurosci. 2021, 15. [Google Scholar] [CrossRef]
- Krüger, M.; Schmid, T.; Krüger, S.; Bober, E.; Braun, T. Functional redundancy of NSCL-1 and NeuroD during development of the petrosal and vestibulocochlear ganglia. Eur. J. Neurosci. 2006, 24, 1581–1590. [Google Scholar] [CrossRef]
- Jia, S.; Ivanov, A.; Blasevic, D.; Müller, T.; Purfürst, B.; Sun, W.; Chen, W.; Poy, M.N.; Rajewsky, N.; Birchmeier, C. Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic β-cell function. Embo. J. 2015, 34, 1417–1433. [Google Scholar] [CrossRef] [Green Version]
- Wiwatpanit, T.; Lorenzen, S.M.; Cantu, J.A.; Foo, C.Z.; Hogan, A.K.; Marquez, F.; Clancy, J.C.; Schipma, M.J.; Cheatham, M.A.; Duggan, A.; et al. Trans-differentiation of outer hair cells into inner hair cells in the absence of INSM1. Nature 2018, 563, 691–695. [Google Scholar] [CrossRef]
- Lorenzen, S.M.; Duggan, A.; Osipovich, A.B.; Magnuson, M.A.; García-Añoveros, J. Insm1 promotes neurogenic proliferation in delaminated otic progenitors. Mech. Dev. 2015, 138, 233–245. [Google Scholar] [CrossRef]
- Wang, V.Y.; Hassan, B.A.; Bellen, H.J.; Zoghbi, H.Y. Drosophila atonal fully rescues the phenotype of Math1 null mice: New functions evolve in new cellular contexts. Curr. Biol. 2002, 12, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Fritzsch, B.; Elliott, K.L. Gene, cell, and organ multiplication drives inner ear evolution. Dev. Biol. 2017, 431, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Jahan, I.; Pan, N.; Elliott, K.L.; Fritzsch, B. The quest for restoring hearing: Understanding ear development more completely. Bioessays 2015, 37, 1016–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzsch, B.; Elliott, K.L. Auditory Nomenclature: Combining Name Recognition with Anatomical Description. Front. Neuroanat. 2018, 12, 99. [Google Scholar] [CrossRef]
- Herranen, A.; Ikäheimo, K.; Lankinen, T.; Pakarinen, E.; Fritzsch, B.; Saarma, M.; Lindahl, M.; Pirvola, U. Deficiency of the ER-stress-regulator MANF triggers progressive outer hair cell death and hearing loss. Cell Death Dis. 2020, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Kempfle, J.S.; Turban, J.L.; Edge, A.S. Sox2 in the differentiation of cochlear progenitor cells. Sci. Rep. 2016, 6, 23293. [Google Scholar] [CrossRef] [Green Version]
- Steevens, A.R.; Glatzer, J.C.; Kellogg, C.C.; Low, W.C.; Santi, P.A.; Kiernan, A.E. SOX2 is required for inner ear growth and cochlear nonsensory formation before sensory development. Development 2019, 146, dev170522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Xu, J.; Xu, P.X. EYA1 and SIX1 drive the neuronal developmental program in cooperation with the SWI/SNF chromatin-remodeling complex and SOX2 in the mammalian inner ear. Development 2012, 139, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Wong, E.Y.; Sun, J.; Xu, J.; Wang, F.; Xu, P.-X. Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev. Cell 2012, 22, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Kempfle, J.S.; Edge, A.S. Pax2 and Sox2 Cooperate to Promote Hair Cell Fate in Inner Ear Stem Cells. Otolaryngol. Head Neck Surg. 2014, 151, P221. [Google Scholar] [CrossRef]
- Mann, Z.F.; Galvez, H.; Pedreno, D.; Chen, Z.; Chrysostomou, E.; Żak, M.; Kang, M.; Canden, E.; Daudet, N. Shaping of inner ear sensory organs through antagonistic interactions between Notch signalling and Lmx1a. Elife 2017, 6, e33323. [Google Scholar] [CrossRef] [Green Version]
- Nichols, D.; Bouma, J.; Kopecky, B.; Jahan, I.; Beisel, K.W.; He, D.; Liu, D.; Fritzsch, B. Interaction with ectopic cochlear crista sensory epithelium disrupts basal cochlear sensory epithelium development in Lmx1a mutant mice. Cell Tissue Res. 2020, in press. [Google Scholar] [CrossRef]
- Huang, Y.; Hill, J.; Yatteau, A.; Wong, L.; Jiang, T.; Petrovic, J.; Gan, L.; Dong, L.; Wu, D.K. Reciprocal negative regulation between Lmx1a and Lmo4 is required for inner ear formation. J. Neurosci. 2018, 38, 5429–5440. [Google Scholar] [CrossRef]
- Pauley, S.; Lai, E.; Fritzsch, B. Foxg1 is required for morphogenesis and histogenesis of the mammalian inner ear. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2006, 235, 2470–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Meng, W.; Kong, W.; He, Z.; Chai, R. The Role of FoxG1 in the Inner Ear. Front. Cell Dev. Biol. 2020, 8, 1539. [Google Scholar] [CrossRef] [PubMed]
- Pirvola, U.; Ylikoski, J.; Trokovic, R.; Hébert, J.M.; McConnell, S.K.; Partanen, J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron 2002, 35, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Roccio, M.; Perny, M.; Ealy, M.; Widmer, H.R.; Heller, S.; Senn, P. Molecular characterization and prospective isolation of human fetal cochlear hair cell progenitors. Nat. Commun. 2018, 9, 4027. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, N.A.; Hassan, B.A.; Wang, V.Y.; Fernandez, M.; Banfi, S.; Bellen, H.J.; Fritzsch, B.; Zoghbi, H.Y. Proprioceptor pathway development is dependent on Math1. Neuron 2001, 30, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.S.; Dymecki, S.M. Rautenlippe Redux—toward a unified view of the precerebellar rhombic lip. Curr. Opin. Cell Biol. 2009, 21, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farago, A.F.; Awatramani, R.B.; Dymecki, S.M. Assembly of the brainstem cochlear nuclear complex is revealed by intersectional and subtractive genetic fate maps. Neuron 2006, 50, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maricich, S.M.; Xia, A.; Mathes, E.L.; Wang, V.Y.; Oghalai, J.S.; Fritzsch, B.; Zoghbi, H.Y. Atoh1-lineal neurons are required for hearing and for the survival of neurons in the spiral ganglion and brainstem accessory auditory nuclei. J. Neurosci. 2009, 29, 11123–11133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Kardon, A.P.; Snyder, L.M.; Kuzirian, M.S.; Minestro, S.; de Souza, L.; Rubio, M.E.; Maricich, S.M.; Ross, S.E. Bhlhb5:: Flpo allele uncovers a requirement for Bhlhb5 for the development of the dorsal cochlear nucleus. Dev. Biol. 2016, 414, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskusnykh, I.Y.; Steshina, E.Y.; Chizhikov, V.V. Loss of Ptf1a leads to a widespread cell-fate misspecification in the brainstem, affecting the development of somatosensory and viscerosensory nuclei. J. Neurosci. 2016, 36, 2691–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bonito, M.; Studer, M.; Puelles, L. Nuclear derivatives and axonal projections originating from rhombomere 4 in the mouse hindbrain. Brain Struct. Funct. 2017, 222, 3509–3542. [Google Scholar] [CrossRef] [Green Version]
- Glover, J.C.; Elliott, K.L.; Erives, A.; Chizhikov, V.V.; Fritzsch, B. Wilhelm His’ lasting insights into hindbrain and cranial ganglia development and evolution. Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lunde, A.; Okaty, B.W.; Dymecki, S.M.; Glover, J.C. Molecular profiling defines evolutionarily conserved transcription factor signatures of major vestibulospinal neuron groups. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Elliott, K.L.; Kersigo, J.; Pan, N.; Jahan, I.; Fritzsch, B. Spiral Ganglion Neuron Projection Development to the Hindbrain in Mice Lacking Peripheral and/or Central Target Differentiation. Front. Neural Circuits 2017, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Guillermo, B. Uncovering the Interplay between Call Fate Specification and Progenitor Dynamics during the Development of the Lower Rhombic Lip. Ph.D. Thesis, Universitat Pompeu Fabra, Barcelona, Spain, 2019. [Google Scholar]
- Hernandez-Miranda, L.R.; Müller, T.; Birchmeier, C. The dorsal spinal cord and hindbrain: From developmental mechanisms to functional circuits. Dev. Biol. 2017, 432, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Kersigo, J.; Gu, L.; Xu, L.; Pan, N.; Vijayakuma, S.; Jones, T.; Shibata, S.B.; Fritzsch, B.; Hansen, M.R. Effects of Neurod1 Expression on Mouse and Human Schwannoma Cells. Laryngoscope 2021, 131, E259–E270. [Google Scholar] [CrossRef]
- Lai, H.C.; Seal, R.P.; Johnson, J.E. Making sense out of spinal cord somatosensory development. Development 2016, 143, 3434–3448. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, K.; Narita, Y.; Fadok, J.; Ducret, S.; Loche, A.; Kitazawa, T.; Genoud, C.; Di Meglio, T.; Thierry, R.; Bacelo, J. Hox2 genes are required for tonotopic map precision and sound discrimination in the mouse auditory brainstem. Cell Rep. 2017, 18, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheah, K.S.; Xu, P.-X. SOX2 in Neurosensory Fate Determination and Differentiation in the Inner Ear. In Sox2; Elsevier: Amsterdam, The Netherlands, 2016; pp. 263–280. [Google Scholar]
- Kondoh, H.; Lovell-Badge, R. Sox2: Biology and Role in Development and Disease; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Kageyama, R.; Shimojo, H.; Ohtsuka, T. Dynamic control of neural stem cells by bHLH factors. Neurosci. Res. 2019, 138, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xu, J.; Xu, P.X. Eya2 expression during mouse embryonic development revealed by Eya2 lacZ knockin reporter and homozygous mice show mild hearing loss. Dev. Dyn. 2021. [CrossRef]
- Duncan, J.S.; Fritzsch, B.; Houston, D.W.; Ketchum, E.M.; Kersigo, J.; Deans, M.R.; Elliott, K.L. Topologically correct central projections of tetrapod inner ear afferents require Fzd3. Sci. Rep. 2019, 9, 10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.; Fritzsch, B. Npr2 null mutants show initial overshooting followed by reduction of spiral ganglion axon projections combined with near-normal cochleotopic projection. Cell Tissue Res. 2019, 378, 15–32. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atoh1+/+ | Atoh1−/− | Neurog−/− | Neurog1–/–/Atoh1−/− | Neurod1−/− | Neurod1–/–/Atoh1−/− | |
|---|---|---|---|---|---|---|
| Matei et al. 2005 | 5.77 (100%) | 5.40 (93%) | 3.08 (53%) | 3.00 (52%) | ||
| Jahan et al. 2010 | 5.90 ± 0.4 | 2.45 ± 0.3 | 2.69 ± 0.1 | |||
| Filova et al. 2020 | 4.6 (E16.5) | 4.5 (E16.5) | 3.1 (E16.5) | 2.4 (E16.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elliott, K.L.; Pavlínková, G.; Chizhikov, V.V.; Yamoah, E.N.; Fritzsch, B. Development in the Mammalian Auditory System Depends on Transcription Factors. Int. J. Mol. Sci. 2021, 22, 4189. https://doi.org/10.3390/ijms22084189
Elliott KL, Pavlínková G, Chizhikov VV, Yamoah EN, Fritzsch B. Development in the Mammalian Auditory System Depends on Transcription Factors. International Journal of Molecular Sciences. 2021; 22(8):4189. https://doi.org/10.3390/ijms22084189
Chicago/Turabian StyleElliott, Karen L., Gabriela Pavlínková, Victor V. Chizhikov, Ebenezer N. Yamoah, and Bernd Fritzsch. 2021. "Development in the Mammalian Auditory System Depends on Transcription Factors" International Journal of Molecular Sciences 22, no. 8: 4189. https://doi.org/10.3390/ijms22084189