
RAD51 Inhibition Induces R-Loop Formation in Early G1 Phase of the Cell Cycle


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. RAD51 Inhibition Induces the Formation of R-Loops
2.2. B02-Induced R-Loops Occur in Early G1 Phase of the Cell Cycle
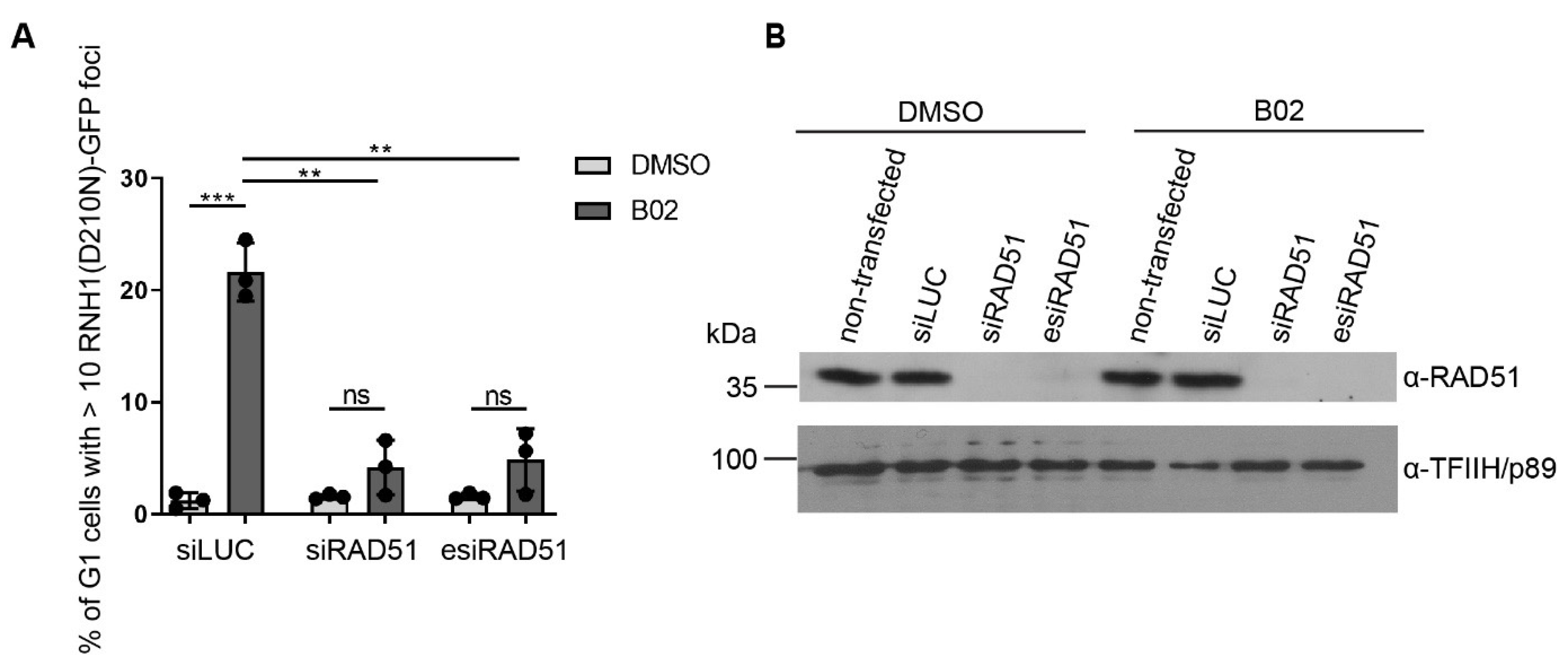
2.3. Formation of B02-Induced R-Loops Requires RAD51
2.4. Inhibition of Transcription Initiation Suppresses Formation of B02-Induced R-Loops
2.5. B02-Induced R-Loops Form at Transcription Start Site of rDNA Locus
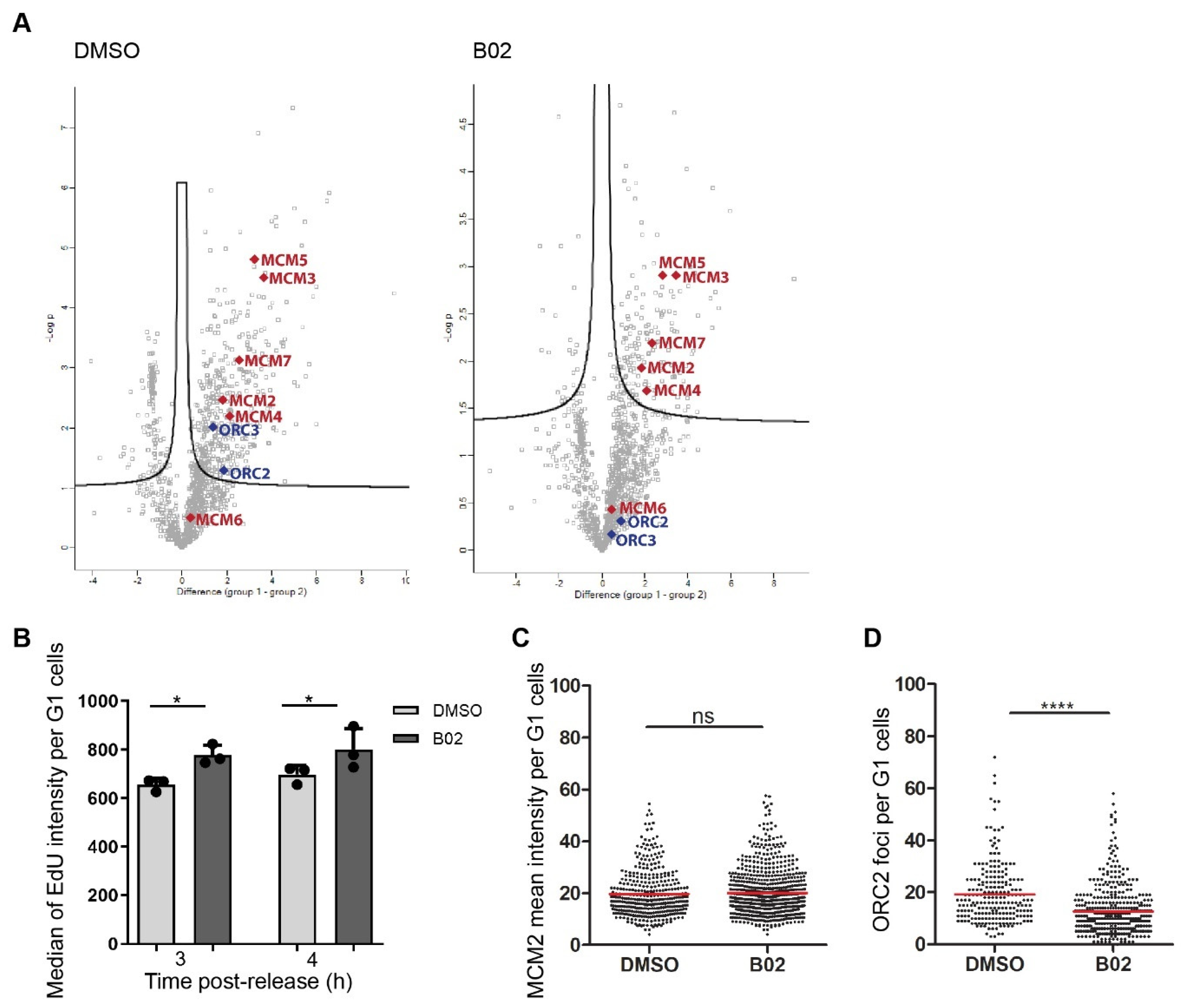
2.6. B02 Induces DNA Synthesis in Early G1 Phase
3. Experimental Procedures
3.1. Cell Culture and Chemicals
3.2. Generation of U-2-OS T-REx RNH1(D210N)-BioID2 Cell Line
3.3. Immunofluorescence Assay
3.4. Detection of RNA: DNA Hybrids with S9.6 Antibody
3.5. Small-Interfering RNA Transfections
3.6. Sample Preparation for Flow Cytometry Measurement
3.7. Preparation of Cell Extracts and Western Blot Analysis
3.8. Chromatin Immunoprecipitation and qPCR
3.9. Immunoprecipitation of Biotinylated Proteins
3.10. Protein Digestion
3.11. nLC-MS 2 Analysis
3.12. Data Analysis
3.13. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, M.; White, R.L.; Davis, R.W. Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feretzaki, M.; Pospisilova, M.; Valador Fernandes, R.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Boque-Sastre, R.; Soler, M.; Oliveira-Mateos, C.; Portela, A.; Moutinho, C.; Sayols, S.; Villanueva, A.; Esteller, M.; Guil, S. Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. Proc. Natl. Acad. Sci. USA 2015, 112, 5785–5790. [Google Scholar] [CrossRef] [Green Version]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair Amst. 2014, 19, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Cerritelli, S.M.; Crouch, R.J. Ribonuclease H: The enzymes in eukaryotes. FEBS J. 2009, 276, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Nowotny, M.; Gaidamakov, S.A.; Ghirlando, R.; Cerritelli, S.M.; Crouch, R.J.; Yang, W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: Insight into HIV reverse transcription. Mol. Cell 2007, 28, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Albulescu, L.O.; Cristini, A.; Gromak, N. Senataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration. J. Mol. Biol. 2017, 429, 3181–3195. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, M.; Clikeman, J.A.; Bates, D.B.; Kogoma, T. RecA protein-dependent R-loop formation in vitro. Genes Dev. 2000, 14, 360–365. [Google Scholar]
- Zaitsev, E.N.; Kowalczykowski, S.C. A novel pairing process promoted by Escherichia coli RecA protein: Inverse DNA and RNA strand exchange. Genes Dev. 2000, 14, 740–749. [Google Scholar]
- Huang, F.; Motlekar, N.A.; Burgwin, C.M.; Napper, A.D.; Diamond, S.L.; Mazin, A.V. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011, 6, 628–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Mazin, A.V. A small molecule inhibitor of human RAD51 potentiates breast cancer cell killing by therapeutic agents in mouse xenografts. PLoS ONE 2014, 9, e100993. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Urban, V.; Dobrovolna, J.; Huhn, D.; Fryzelkova, J.; Bartek, J.; Janscak, P. RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 2016, 214, 401–415. [Google Scholar] [CrossRef] [Green Version]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 2017, 65, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Teloni, F.; Michelena, J.; Lezaja, A.; Kilic, S.; Ambrosi, C.; Menon, S.; Dobrovolna, J.; Imhof, R.; Janscak, P.; Baubec, T.; et al. Efficient Pre-mRNA Cleavage Prevents Replication-Stress-Associated Genome Instability. Mol. Cell 2019, 73, 670–683. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Lima, W.F.; Crooke, S.T. Investigating the structure of human RNase H1 by site-directed mutagenesis. J. Biol. Chem. 2001, 276, 23547–23553. [Google Scholar] [CrossRef] [Green Version]
- Tsuzuki, T.; Fujii, Y.; Sakumi, K.; Tominaga, Y.; Nakao, K.; Sekiguchi, M.; Matsushiro, A.; Yoshimura, Y.; Morita, T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6236–6240. [Google Scholar] [CrossRef] [Green Version]
- Tazi, J.; Bakkour, N.; Soret, J.; Zekri, L.; Hazra, B.; Laine, W.; Baldeyrou, B.; Lansiaux, A.; Bailly, C. Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol. Pharmacol. 2005, 67, 1186–1194. [Google Scholar] [CrossRef]
- Tuduri, S.; Crabbe, L.; Conti, C.; Tourriere, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Aubry, A.; Pearson, J.D.; Huang, K.; Livne-Bar, I.; Ahmad, M.; Jagadeesan, M.; Khetan, V.; Ketela, T.; Brown, K.R.; Yu, T.; et al. Functional genomics identifies new synergistic therapies for retinoblastoma. Oncogene 2020, 39, 5338–5357. [Google Scholar] [CrossRef]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Dumelie, J.G.; Jaffrey, S.R. Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Ratmeyer, L.; Vinayak, R.; Zhong, Y.Y.; Zon, G.; Wilson, W.D. Sequence specific thermodynamic and structural properties for DNA.RNA duplexes. Biochemistry 1994, 33, 5298–5304. [Google Scholar] [CrossRef]
- El Hage, A.; Tollervey, D. Immunoprecipitation of RNA:DNA Hybrids from Budding Yeast. Methods Mol. Biol. 2018, 1703, 109–129. [Google Scholar] [CrossRef]
- Chen, J.Y.; Zhang, X.; Fu, X.D.; Chen, L. R-ChIP for genome-wide mapping of R-loops by using catalytically inactive RNASEH1. Nat. Protoc. 2019, 14, 1661–1685. [Google Scholar] [CrossRef]
- Donovan, S.; Harwood, J.; Drury, L.S.; Diffley, J.F. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 5611–5616. [Google Scholar] [CrossRef] [Green Version]
- Evrin, C.; Clarke, P.; Zech, J.; Lurz, R.; Sun, J.; Uhle, S.; Li, H.; Stillman, B.; Speck, C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 2009, 106, 20240–20245. [Google Scholar] [CrossRef] [Green Version]
- Remus, D.; Beuron, F.; Tolun, G.; Griffith, J.D.; Morris, E.P.; Diffley, J.F. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009, 139, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, K.; On, K.F.; Diffley, J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Ticau, S.; Friedman, L.J.; Ivica, N.A.; Gelles, J.; Bell, S.P. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 2015, 161, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Marti, T.M.; Hefner, E.; Feeney, L.; Natale, V.; Cleaver, J.E. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2006, 103, 9891–9896. [Google Scholar] [CrossRef] [Green Version]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascakova, Z.; Boleslavska, B.; Urban, V.; Oravetzova, A.; Vlachova, E.; Janscak, P.; Dobrovolna, J. RAD51 Inhibition Induces R-Loop Formation in Early G1 Phase of the Cell Cycle. Int. J. Mol. Sci. 2021, 22, 3740. https://doi.org/10.3390/ijms22073740
Nascakova Z, Boleslavska B, Urban V, Oravetzova A, Vlachova E, Janscak P, Dobrovolna J. RAD51 Inhibition Induces R-Loop Formation in Early G1 Phase of the Cell Cycle. International Journal of Molecular Sciences. 2021; 22(7):3740. https://doi.org/10.3390/ijms22073740
Chicago/Turabian StyleNascakova, Zuzana, Barbora Boleslavska, Vaclav Urban, Anna Oravetzova, Edita Vlachova, Pavel Janscak, and Jana Dobrovolna. 2021. "RAD51 Inhibition Induces R-Loop Formation in Early G1 Phase of the Cell Cycle" International Journal of Molecular Sciences 22, no. 7: 3740. https://doi.org/10.3390/ijms22073740