
Bordetella Adenylate Cyclase Toxin Elicits Airway Mucin Secretion through Activation of the cAMP Response Element Binding Protein


 , , , ,
, , , ,  and
and 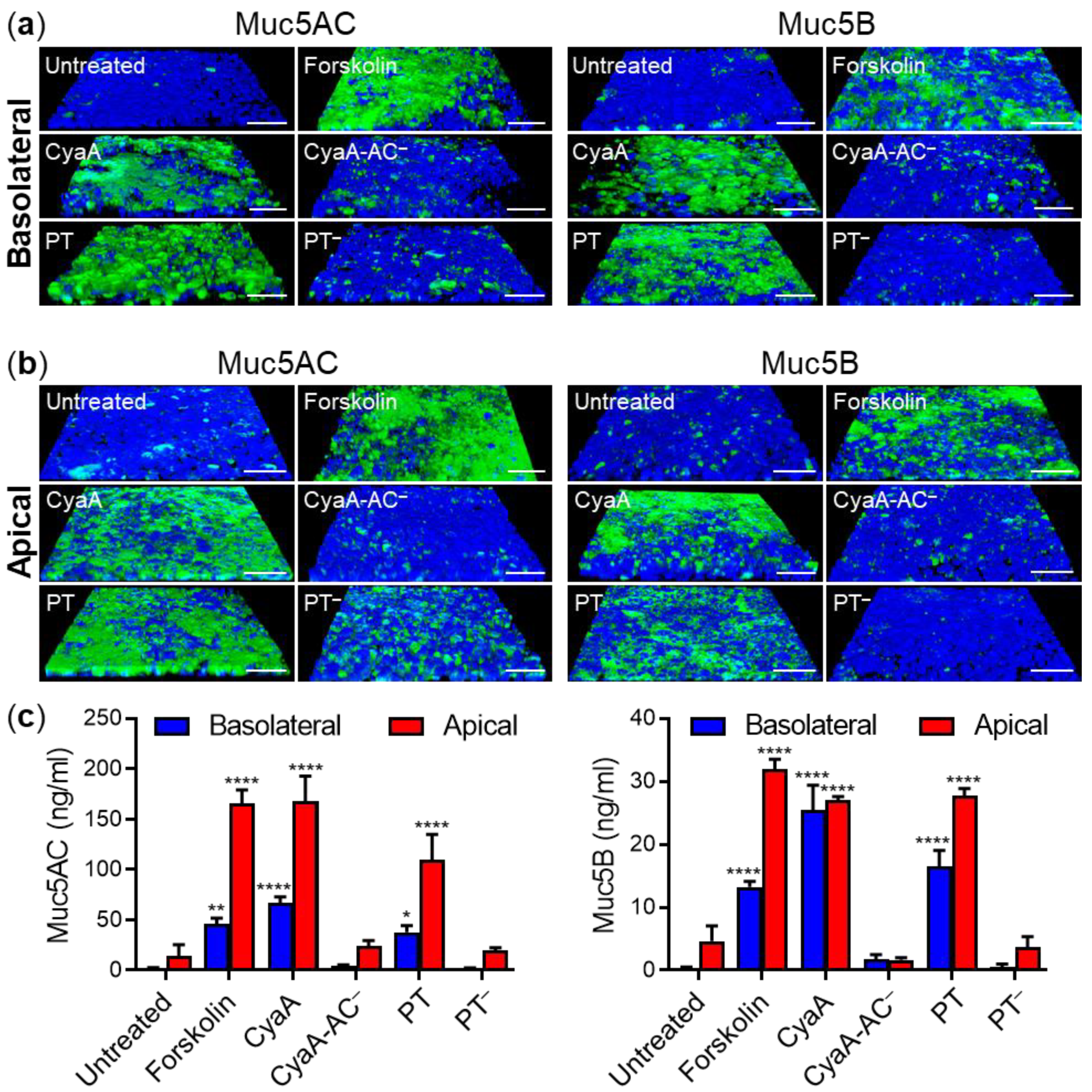{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Both CyaA and PT Trigger Mucin Production by Pseudostratified Airway Epithelial Cells
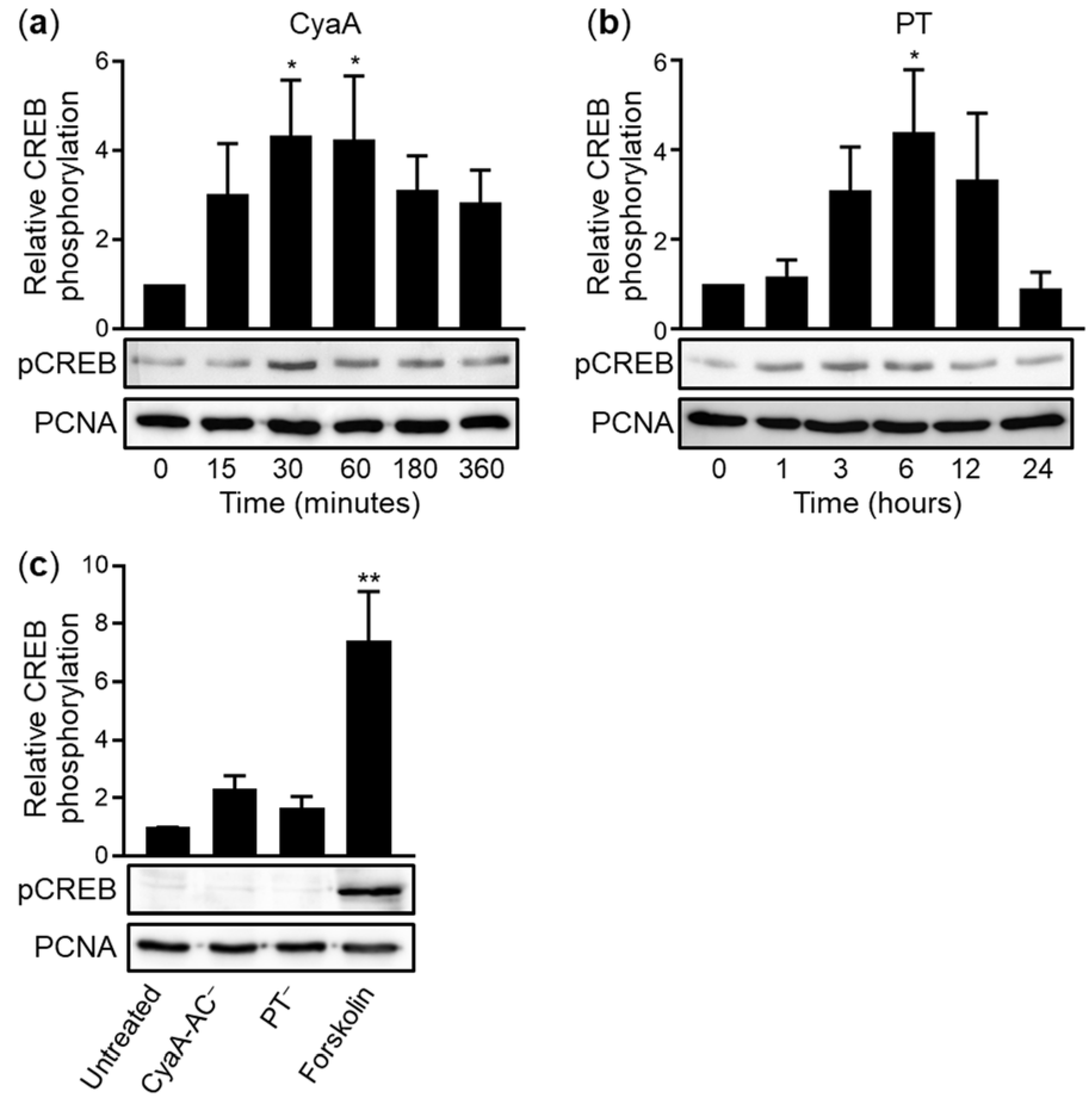
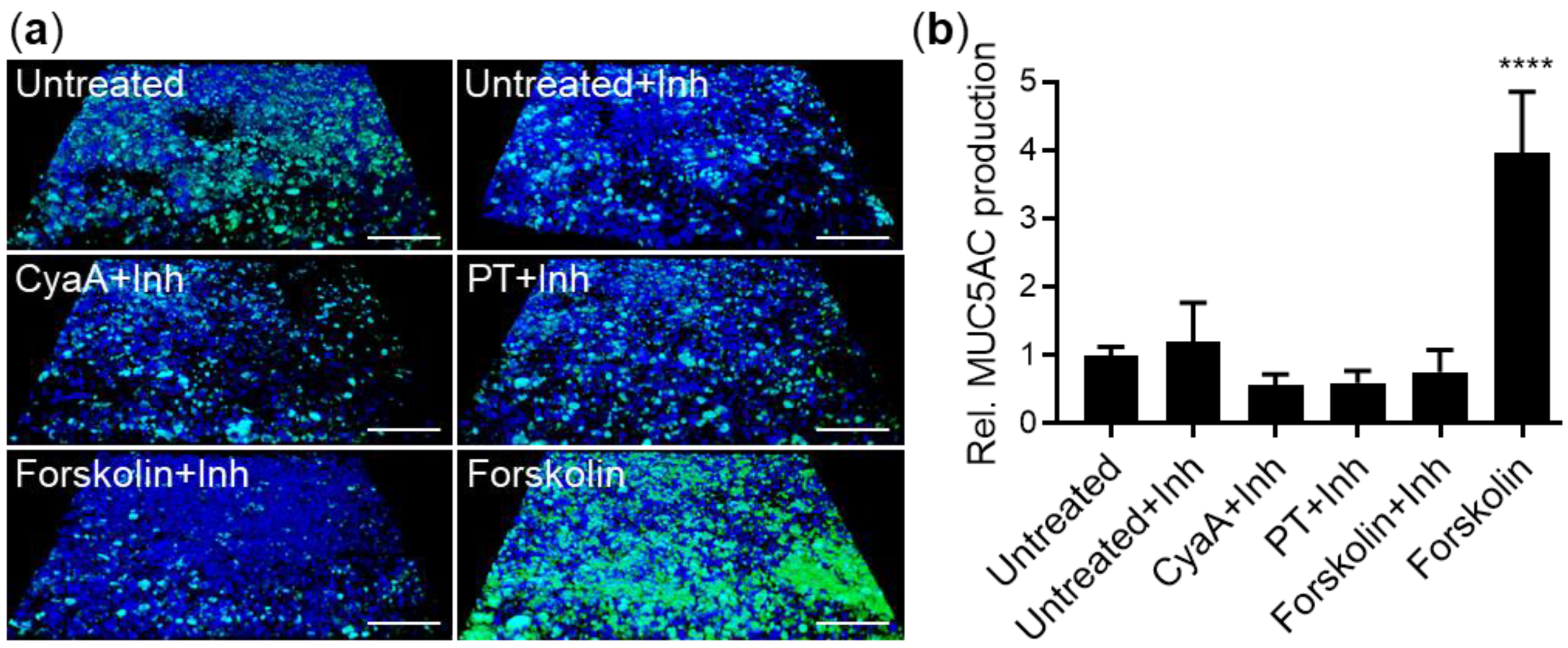
2.2. CyaA and PT Trigger Phosphorylation of CREB in VA10 Cell Layers
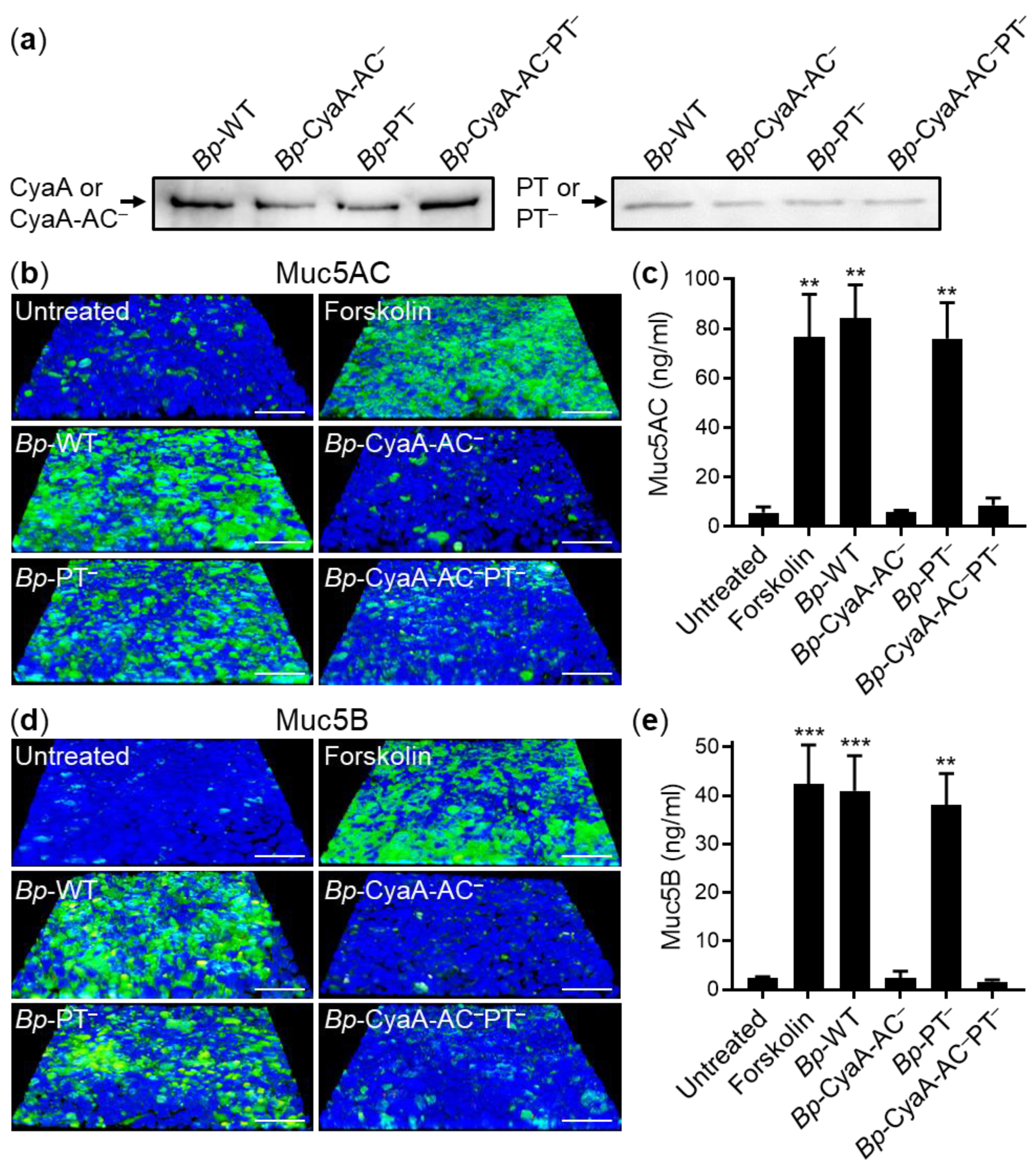
2.3. Only Secretion of CyaA Triggers Mucin Production in B. pertussis-Infected VA10 Cell Layers and in Infected Mouse Tracheas In Vivo
3. Discussion
4. Materials and Methods
4.1. VA10 Cell Layers
4.2. Intoxication Experiments
4.3. Confocal Microscopy
4.4. Muc5AC and Muc5B ELISA
4.5. Immunodetection of Phosphorylated CREB
4.6. B. pertussis Strains
4.7. Immunodetection of the CyaA and PT Toxins and Toxoids
4.8. Bacterial Infection of VA10 Cell Layers
4.9. Mice Infections and Ethics Statement
4.10. Staining and Quantification of Tracheal Mucins
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Mattoo, S.; Cherry, J.D. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Althouse, B.M.; Scarpino, S.V. Asymptomatic transmission and the resurgence of Bordetella pertussis. BMC Med. 2015, 13, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenech de Celles, M.; Magpantay, F.M.; King, A.A.; Rohani, P. The pertussis enigma: Reconciling epidemiology, immunology and evolution. Proc. Biol. Sci. 2016, 283, 20152309. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.J.; Gunning, C.E.; MacLeod, W.B.; Mwananyanda, L.; Thea, D.M.; Pieciak, R.C.; Kwenda, G.; Mupila, Z.; Rohani, P. Asymptomatic Bordetella pertussis infections in a longitudinal cohort of young African infants and their mothers. eLife 2021, 10, e65663. [Google Scholar] [CrossRef]
- Yeung, K.H.T.; Duclos, P.; Nelson, E.A.S.; Hutubessy, R.C.W. An update of the global burden of pertussis in children younger than 5 years: A modelling study. Lancet Infect. Dis. 2017, 17, 974–980. [Google Scholar] [CrossRef]
- Scanlon, K.; Skerry, C.; Carbonetti, N. Role of Major Toxin Virulence Factors in Pertussis Infection and Disease Pathogenesis. Adv. Exp. Med. Biol. 2019, 1183, 35–51. [Google Scholar] [CrossRef]
- Linhartova, I.; Bumba, L.; Masin, J.; Basler, M.; Osicka, R.; Kamanova, J.; Prochazkova, K.; Adkins, I.; Hejnova-Holubova, J.; Sadilkova, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, M.S.; Weiss, A.A. Adenylate cyclase toxin is critical for colonization and pertussis toxin is critical for lethal infection by Bordetella pertussis in infant mice. Infect. Immun. 1990, 58, 3445–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiso, N.; Rocancourt, M.; Szatanik, M.; Alonso, J.M. Bordetella adenylate cyclase is a virulence associated factor and an immunoprotective antigen. Microb. Pathog. 1989, 7, 373–380. [Google Scholar] [CrossRef]
- Skopova, K.; Tomalova, B.; Kanchev, I.; Rossmann, P.; Svedova, M.; Adkins, I.; Bibova, I.; Tomala, J.; Masin, J.; Guiso, N.; et al. Cyclic AMP-Elevating Capacity of Adenylate Cyclase Toxin-Hemolysin Is Sufficient for Lung Infection but Not for Full Virulence of Bordetella pertussis. Infect. Immun. 2017, 85, e00937-16. [Google Scholar] [CrossRef] [Green Version]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73, ftv075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, J.; Cerny, O.; Osickova, A.; Linhartova, I.; Masin, J.; Bumba, L.; Sebo, P.; Osicka, R. Structure-Function Relationships Underlying the Capacity of Bordetella Adenylate Cyclase Toxin to Disarm Host Phagocytes. Toxins 2017, 9, 300. [Google Scholar] [CrossRef]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Morova, J.; Osicka, R.; Masin, J.; Sebo, P. RTX cytotoxins recognize beta2 integrin receptors through N-linked oligosaccharides. Proc. Natl. Acad. Sci. USA 2008, 105, 5355–5360. [Google Scholar] [CrossRef] [Green Version]
- Osicka, R.; Osickova, A.; Hasan, S.; Bumba, L.; Cerny, J.; Sebo, P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. eLife 2015, 4, e10766. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Osickova, A.; Bumba, L.; Novak, P.; Sebo, P.; Osicka, R. Interaction of Bordetella adenylate cyclase toxin with complement receptor 3 involves multivalent glycan binding. FEBS Lett. 2015, 589, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Wald, T.; Osickova, A.; Masin, J.; Liskova, P.M.; Petry-Podgorska, I.; Matousek, T.; Sebo, P.; Osicka, R. Transmembrane segments of complement receptor 3 do not participate in cytotoxic activities but determine receptor structure required for action of Bordetella adenylate cyclase toxin. Pathog. Dis. 2016, 74, ftw008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masin, J.; Osickova, A.; Jurnecka, D.; Klimova, N.; Khaliq, H.; Sebo, P.; Osicka, R. Retargeting from the CR3 to the LFA-1 receptor uncovers the adenylyl cyclase enzyme-translocating segment of Bordetella adenylate cyclase toxin. J. Biol. Chem. 2020, 295, 9349–9365. [Google Scholar] [CrossRef] [PubMed]
- Osickova, A.; Masin, J.; Fayolle, C.; Krusek, J.; Basler, M.; Pospisilova, E.; Leclerc, C.; Osicka, R.; Sebo, P. Adenylate cyclase toxin translocates across target cell membrane without forming a pore. Mol. Microbiol. 2010, 75, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Cook, G.H.; Goldhammer, A.R.; Berkowitz, S.A. Calmodulin activates prokaryotic adenylate cyclase. Proc. Natl. Acad. Sci. USA 1980, 77, 3841–3844. [Google Scholar] [CrossRef] [Green Version]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.C.; Gray, M.C.; Hewlett, E.L. Cyclic AMP-mediated suppression of neutrophil extracellular trap formation and apoptosis by the Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 2014, 82, 5256–5269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerny, O.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Sebo, P. cAMP Signaling of Adenylate Cyclase Toxin Blocks the Oxidative Burst of Neutrophils through Epac-Mediated Inhibition of Phospholipase C Activity. J. Immunol. 2017, 198, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Kamanova, J.; Kofronova, O.; Masin, J.; Genth, H.; Vojtova, J.; Linhartova, I.; Benada, O.; Just, I.; Sebo, P. Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 2008, 181, 5587–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, S.; Rahman, W.U.; Sebo, P.; Osicka, R. Distinct Spatiotemporal Distribution of Bacterial Toxin-Produced Cellular cAMP Differentially Inhibits Opsonophagocytic Signaling. Toxins 2019, 11, 362. [Google Scholar] [CrossRef] [Green Version]
- Gueirard, P.; Druilhe, A.; Pretolani, M.; Guiso, N. Role of adenylate cyclase-hemolysin in alveolar macrophage apoptosis during Bordetella pertussis infection in vivo. Infect. Immun. 1998, 66, 1718–1725. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, J.N.; Cerny, O.; Linhartova, I.; Masin, J.; Osicka, R.; Sebo, P. cAMP signalling of Bordetella adenylate cyclase toxin through the SHP-1 phosphatase activates the BimEL-Bax pro-apoptotic cascade in phagocytes. Cell. Microbiol. 2016, 18, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, J.N.; Holubova, J.; Benada, O.; Kofronova, O.; Stehlik, L.; Vasakova, M.; Sebo, P. Bordetella Adenylate Cyclase Toxin Inhibits Monocyte-to-Macrophage Transition and Dedifferentiates Human Alveolar Macrophages into Monocyte-like Cells. mBio 2019, 10, e01743-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, J.N.; Sebo, P. Adenylate Cyclase Toxin Tinkering With Monocyte-Macrophage Differentiation. Front. Immunol. 2020, 11, 2181. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.M.; Young, W.W., Jr.; Lechler, S.M.; Gray, M.C.; Leppla, S.H.; Hewlett, E.L. Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J. Biol. Chem. 1989, 264, 14792–14796. [Google Scholar] [CrossRef]
- Sheppard, D. Airway epithelial integrins: Why so many? Am. J. Respir. Cell Mol. Biol. 1998, 19, 349–351. [Google Scholar] [CrossRef]
- Sheppard, D. Functions of pulmonary epithelial integrins: From development to disease. Physiol. Rev. 2003, 83, 673–686. [Google Scholar] [CrossRef] [Green Version]
- Eby, J.C.; Gray, M.C.; Warfel, J.M.; Paddock, C.D.; Jones, T.F.; Day, S.R.; Bowden, J.; Poulter, M.D.; Donato, G.M.; Merkel, T.J.; et al. Quantification of the adenylate cyclase toxin of Bordetella pertussis in vitro and during respiratory infection. Infect. Immun. 2013, 81, 1390–1398. [Google Scholar] [CrossRef] [Green Version]
- Locht, C.; Coutte, L.; Mielcarek, N. The ins and outs of pertussis toxin. FEBS J. 2011, 278, 4668–4682. [Google Scholar] [CrossRef]
- Armstrong, G.D.; Howard, L.A.; Peppler, M.S. Use of glycosyltransferases to restore pertussis toxin receptor activity to asialoagalactofetuin. J. Biol. Chem. 1988, 263, 8677–8684. [Google Scholar] [CrossRef]
- el Baya, A.; Linnemann, R.; von Olleschik-Elbheim, L.; Robenek, H.; Schmidt, M.A. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur. J. Cell Biol. 1997, 73, 40–48. [Google Scholar] [PubMed]
- Kugler, S.; Bocker, K.; Heusipp, G.; Greune, L.; Kim, K.S.; Schmidt, M.A. Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell. Microbiol. 2007, 9, 619–632. [Google Scholar] [CrossRef]
- Plaut, R.D.; Carbonetti, N.H. Retrograde transport of pertussis toxin in the mammalian cell. Cell. Microbiol. 2008, 10, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Cilenti, L.; Taylor, M.; Showman, A.; Tatulian, S.A.; Teter, K. Thermal Unfolding of the Pertussis Toxin S1 Subunit Facilitates Toxin Translocation to the Cytosol by the Mechanism of Endoplasmic Reticulum-Associated Degradation. Infect. Immun. 2016, 84, 3388–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Ernst, K.; Eberhardt, N.; Mittler, A.K.; Sonnabend, M.; Anastasia, A.; Freisinger, S.; Schiene-Fischer, C.; Malesevic, M.; Barth, H. Pharmacological Cyclophilin Inhibitors Prevent Intoxication of Mammalian Cells with Bordetella pertussis Toxin. Toxins 2018, 10, 181. [Google Scholar] [CrossRef] [Green Version]
- Ernst, K.; Mittler, A.K.; Winkelmann, V.; Kling, C.; Eberhardt, N.; Anastasia, A.; Sonnabend, M.; Lochbaum, R.; Wirsching, J.; Sakari, M.; et al. Pharmacological targeting of host chaperones protects from pertussis toxin in vitro and in vivo. Sci. Rep. 2021, 11, 5429. [Google Scholar] [CrossRef] [PubMed]
- Katada, T.; Ui, M. Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein. Proc. Natl. Acad. Sci. USA 1982, 79, 3129–3133. [Google Scholar] [CrossRef] [Green Version]
- Pittman, M. The concept of pertussis as a toxin-mediated disease. Pediatric Infect. Dis. 1984, 3, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Kurose, H. G(i/o) protein-dependent and -independent actions of Pertussis Toxin (PTX). Toxins 2011, 3, 884–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonetti, N.H. Pertussis leukocytosis: Mechanisms, clinical relevance and treatment. Pathog. Dis. 2016, 74, ftw087. [Google Scholar] [CrossRef] [Green Version]
- Paddock, C.D.; Sanden, G.N.; Cherry, J.D.; Gal, A.A.; Langston, C.; Tatti, K.M.; Wu, K.H.; Goldsmith, C.S.; Greer, P.W.; Montague, J.L.; et al. Pathology and pathogenesis of fatal Bordetella pertussis infection in infants. Clin. Infect. Dis. 2008, 47, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, K.; Zipprich, J.; Harriman, K.; Murray, E.L.; Gornbein, J.; Hammer, S.J.; Yeganeh, N.; Adachi, K.; Cherry, J.D. Risk Factors Associated With Infant Deaths From Pertussis: A Case-Control Study. Clin. Infect. Dis. 2015, 61, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Derrien, M.; van Passel, M.W.; van de Bovenkamp, J.H.; Schipper, R.G.; de Vos, W.M.; Dekker, J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samet, J.M.; Cheng, P.W. The role of airway mucus in pulmonary toxicology. Env. Health Perspect. 1994, 102 (Suppl. 2), 89–103. [Google Scholar] [CrossRef] [Green Version]
- van Putten, J.P.M.; Strijbis, K. Transmembrane Mucins: Signaling Receptors at the Intersection of Inflammation and Cancer. J. Innate Immun. 2017, 9, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Andrianifahanana, M.; Moniaux, N.; Batra, S.K. Regulation of mucin expression: Mechanistic aspects and implications for cancer and inflammatory diseases. Biochim. Et Biophys. Acta 2006, 1765, 189–222. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef] [PubMed]
- Belcher, C.E.; Drenkow, J.; Kehoe, B.; Gingeras, T.R.; McNamara, N.; Lemjabbar, H.; Basbaum, C.; Relman, D.A. The transcriptional responses of respiratory epithelial cells to Bordetella pertussis reveal host defensive and pathogen counter-defensive strategies. Proc. Natl. Acad. Sci. USA 2000, 97, 13847–13852. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.; Kulkarni, N.N.; Asbjarnarson, A.; Linhartova, I.; Osicka, R.; Sebo, P.; Gudmundsson, G.H. Bordetella pertussis Adenylate Cyclase Toxin Disrupts Functional Integrity of Bronchial Epithelial Layers. Infect. Immun. 2018, 86, e00445-17. [Google Scholar] [CrossRef] [Green Version]
- Barovsky, K.; Pedone, C.; Brooker, G. Distinct mechanisms of forskolin-stimulated cyclic AMP accumulation and forskolin-potentiated hormone responses in C6-2B cells. Mol. Pharmacol. 1984, 25, 256–260. [Google Scholar]
- Kim, S.W.; Hong, J.S.; Ryu, S.H.; Chung, W.C.; Yoon, J.H.; Koo, J.S. Regulation of mucin gene expression by CREB via a nonclassical retinoic acid signaling pathway. Mol. Cell Biol. 2007, 27, 6933–6947. [Google Scholar] [CrossRef] [Green Version]
- Tyson, D.R.; Swarthout, J.T.; Partridge, N.C. Increased osteoblastic c-fos expression by parathyroid hormone requires protein kinase A phosphorylation of the cyclic adenosine 3′,5′-monophosphate response element-binding protein at serine 133. Endocrinology 1999, 140, 1255–1261. [Google Scholar] [CrossRef]
- Cerny, O.; Kamanova, J.; Masin, J.; Bibova, I.; Skopova, K.; Sebo, P. Bordetella pertussis Adenylate Cyclase Toxin Blocks Induction of Bactericidal Nitric Oxide in Macrophages through cAMP-Dependent Activation of the SHP-1 Phosphatase. J. Immunol. 2015, 194, 4901–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Fouts, T.R.; Lewis, G.K. Pertussis toxin and the adenylate cyclase toxin from Bordetella pertussis activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cAMP-dependent pathway. J. Leukoc. Biol. 2002, 72, 962–969. [Google Scholar]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem. 2015, 58, 5075–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.X.; Gardner, R.; Xue, C.; Qian, D.Z.; Xie, F.; Thomas, G.; Kazmierczak, S.C.; Habecker, B.A.; Xiao, X. Systemic Inhibition of CREB is Well-tolerated in vivo. Sci. Rep. 2016, 6, 34513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogel, A.; Hanski, E. Distinct steps in the penetration of adenylate cyclase toxin of Bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J. Biol. Chem. 1992, 267, 22599–22605. [Google Scholar] [CrossRef]
- Dal Molin, F.; Tonello, F.; Ladant, D.; Zornetta, I.; Zamparo, I.; Di Benedetto, G.; Zaccolo, M.; Montecucco, C. Cell entry and cAMP imaging of anthrax edema toxin. EMBO J. 2006, 25, 5405–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teter, K. Intracellular Trafficking and Translocation of Pertussis Toxin. Toxins 2019, 11, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eby, J.C.; Ciesla, W.P.; Hamman, W.; Donato, G.M.; Pickles, R.J.; Hewlett, E.L.; Lencer, W.I. Selective translocation of the Bordetella pertussis adenylate cyclase toxin across the basolateral membranes of polarized epithelial cells. J. Biol. Chem. 2010, 285, 10662–10670. [Google Scholar] [CrossRef] [Green Version]
- Guevara, C.; Zhang, C.; Gaddy, J.A.; Iqbal, J.; Guerra, J.; Greenberg, D.P.; Decker, M.D.; Carbonetti, N.; Starner, T.D.; McCray, P.B., Jr.; et al. Highly differentiated human airway epithelial cells: A model to study host cell-parasite interactions in pertussis. Infect. Dis. 2016, 48, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Abdullah, L.H.; Doyle, S.P.; Nguyen, K.; Ribeiro, C.M.; Vasquez, P.A.; Forest, M.G.; Lethem, M.I.; Dickey, B.F.; Davis, C.W. Baseline Goblet Cell Mucin Secretion in the Airways Exceeds Stimulated Secretion over Extended Time Periods, and Is Sensitive to Shear Stress and Intracellular Mucin Stores. PLoS ONE 2015, 10, e0127267. [Google Scholar] [CrossRef]
- Stanek, O.; Masin, J.; Osicka, R.; Jurnecka, D.; Osickova, A.; Sebo, P. Rapid Purification of Endotoxin-Free RTX Toxins. Toxins 2019, 11, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osicka, R.; Osickova, A.; Basar, T.; Guermonprez, P.; Rojas, M.; Leclerc, C.; Sebo, P. Delivery of CD8(+) T-cell epitopes into major histocompatibility complex class I antigen presentation pathway by Bordetella pertussis adenylate cyclase: Delineation of cell invasive structures and permissive insertion sites. Infect. Immun. 2000, 68, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizza, M.; Covacci, A.; Bartoloni, A.; Perugini, M.; Nencioni, L.; De Magistris, M.T.; Villa, L.; Nucci, D.; Manetti, R.; Bugnoli, M.; et al. Mutants of pertussis toxin suitable for vaccine development. Science 1989, 246, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-Driven Folding of RTX Domain beta-Rolls Ratchets Translocation of RTX Proteins through Type I Secretion Ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Gray, M.C.; Guo, L.; Sebo, P.; Hewlett, E.L. Epitope mapping of monoclonal antibodies against Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 1999, 67, 2090–2095. [Google Scholar] [CrossRef] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malandra, A.; Rahman, W.U.; Klimova, N.; Streparola, G.; Holubova, J.; Osickova, A.; Bariselli, S.; Sebo, P.; Osicka, R. Bordetella Adenylate Cyclase Toxin Elicits Airway Mucin Secretion through Activation of the cAMP Response Element Binding Protein. Int. J. Mol. Sci. 2021, 22, 9064. https://doi.org/10.3390/ijms22169064
Malandra A, Rahman WU, Klimova N, Streparola G, Holubova J, Osickova A, Bariselli S, Sebo P, Osicka R. Bordetella Adenylate Cyclase Toxin Elicits Airway Mucin Secretion through Activation of the cAMP Response Element Binding Protein. International Journal of Molecular Sciences. 2021; 22(16):9064. https://doi.org/10.3390/ijms22169064
Chicago/Turabian StyleMalandra, Anna, Waheed Ur Rahman, Nela Klimova, Gaia Streparola, Jana Holubova, Adriana Osickova, Simone Bariselli, Peter Sebo, and Radim Osicka. 2021. "Bordetella Adenylate Cyclase Toxin Elicits Airway Mucin Secretion through Activation of the cAMP Response Element Binding Protein" International Journal of Molecular Sciences 22, no. 16: 9064. https://doi.org/10.3390/ijms22169064