3. Materials and Methods
3.1. AlphaScreen Assay
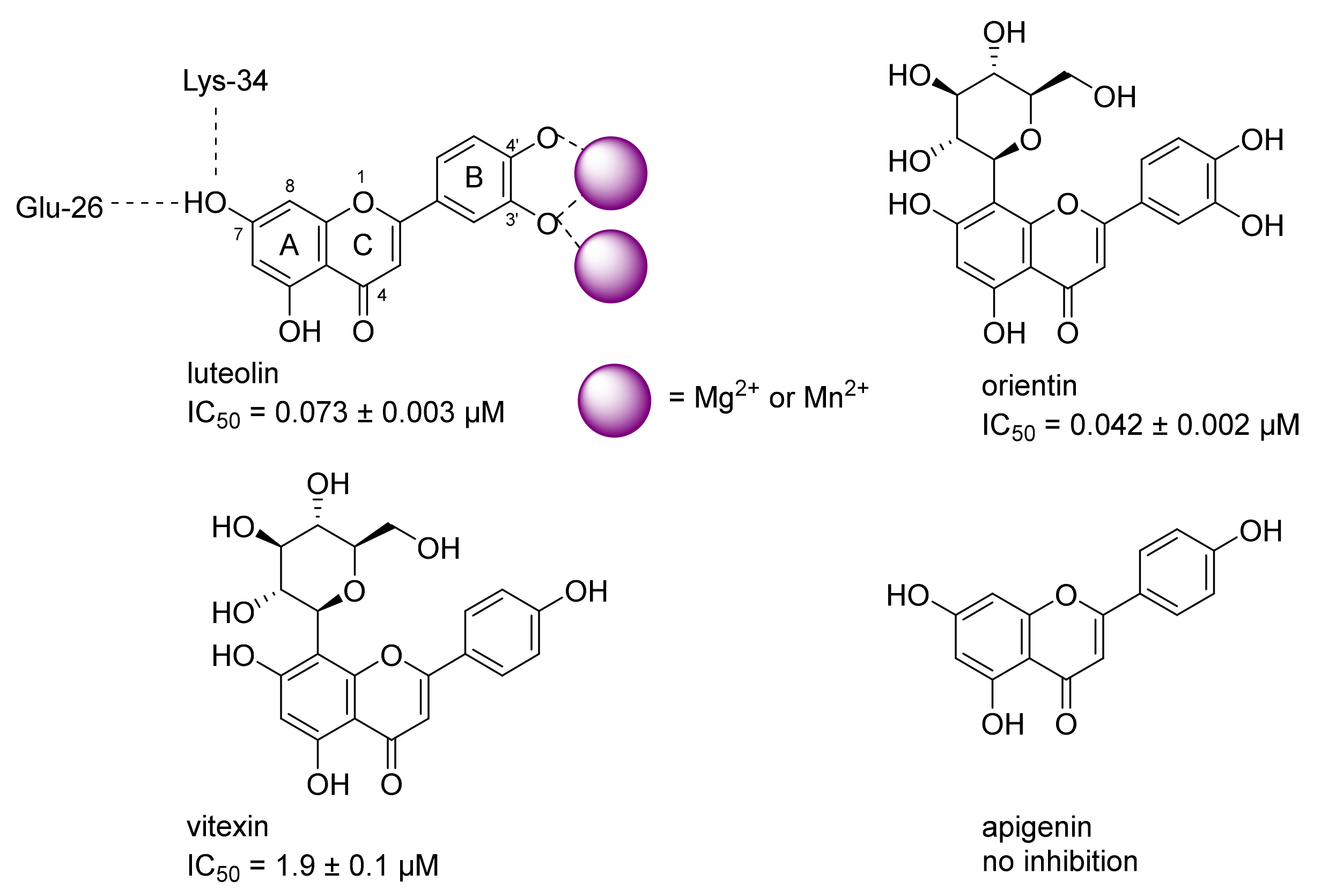
AlphaScreen experiments were performed using a Perkin Elmer Enspire plate reader in 96-well ProxiPlates. Biotinylated L-742.001 derivative [
37] was captured on Streptavidin-coated donor beads (Perkin Elmer). Separately, GST-PA-Nter fusion protein was bound to GSH-coated acceptor beads (Perkin Elmer). These solutions were incubated for 60 min at room temperature in the dark and subsequently mixed and incubated for an additional 120 min. In experiments screening for endonuclease inhibitors, compounds were mixed with both types of beads prior to the 120-min incubation. The optimal concentrations of biotinylated L-742.001 derivative and GST-PA-Nter were 15 nM and 50 nM, respectively. The concentrations of donor and acceptor beads were 5 µg/mL each in a 50 µL reaction volume. All experiments were performed in 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween20, 1 mM MnCl
2, 10 mM MgCl
2, and 1 mM 2-mercaptoethanol.
3.2. Cloning, Expression and Purification of Recombinant Proteins
DNA encoding the first 196 amino acids of the N-terminal domain of the influenza polymerase acidic subunit (PA-Nter) from the viral strain A/California/07/2009 (H1N1) (GenBank accession No. CY121685.1) was prepared using GenScript USA Inx. The flexible loop (residues 51–72) was replaced with a GGS linker [
52]. Constructs with affinity tags (GST-PA-Nter and His
6-SUMO-Nter) were prepared for both wild-type and I 38T PA-Nter. First, DNA encoding PA-Nter was inserted into the plasmid pGEX-1λT. Next, PA-Nter with a (GS)
4 linker and N-terminal extension was cloned into the plasmid pETM11-SUMO3 (EMBL, Heidelberg, Germany) using BamHI and XhoI sites. The I38T mutation was introduced into both GST- and His
6-SUMO-tagged constructs using the following primers for site-directed mutagenesis: 5′–CAAGTTTGCTGCAACATGCACACAT TTG–3′ and 5′–CAAATGTGTGCATGTTGCACAAACTTG–3′. All tagged PA-Nter constructs were expressed in
E. coli BL21 (DE3) RIL. Cells were harvested and resuspended in lysis buffer (25 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA (GST) or 50 mM Tris/HCl, pH 8.0, 200 mM NaCl, 10 mM imidazole (His
6-SUMO)) and lysed with an EmulsiFlex device (Avestin, Ottawa, ON, Canada) at a pressure of 1200 bar. GST-tagged PA-Nter soluble proteins were loaded onto a glutathione-agarose column (ThermoFisher Scientific) and eluted with elution buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 10 mM reduced L glutathione, 1 mM EDTA). Analogously, His
6-SUMO PA-Nter was purified using Ni-NTA Agarose (Roche Diagnostics GmbH, Mannheim, Germany) and eluted with Ni-NTA elution buffer (50 mM Tris/HCl, pH 8.0, 200 mM NaCl, 250 mM imidazole). The His
6-SUMO tag was removed by the ULP1 protease. All proteins were purified on a Superdex 75 (GE Healthcare/Amersham Pharmacia, Uppsala, Sweden) gel filtration chromatography column, yielding >95% purity as estimated by SDS-PAGE.
3.3. Gel-Based Endonuclease Inhibitory Assay
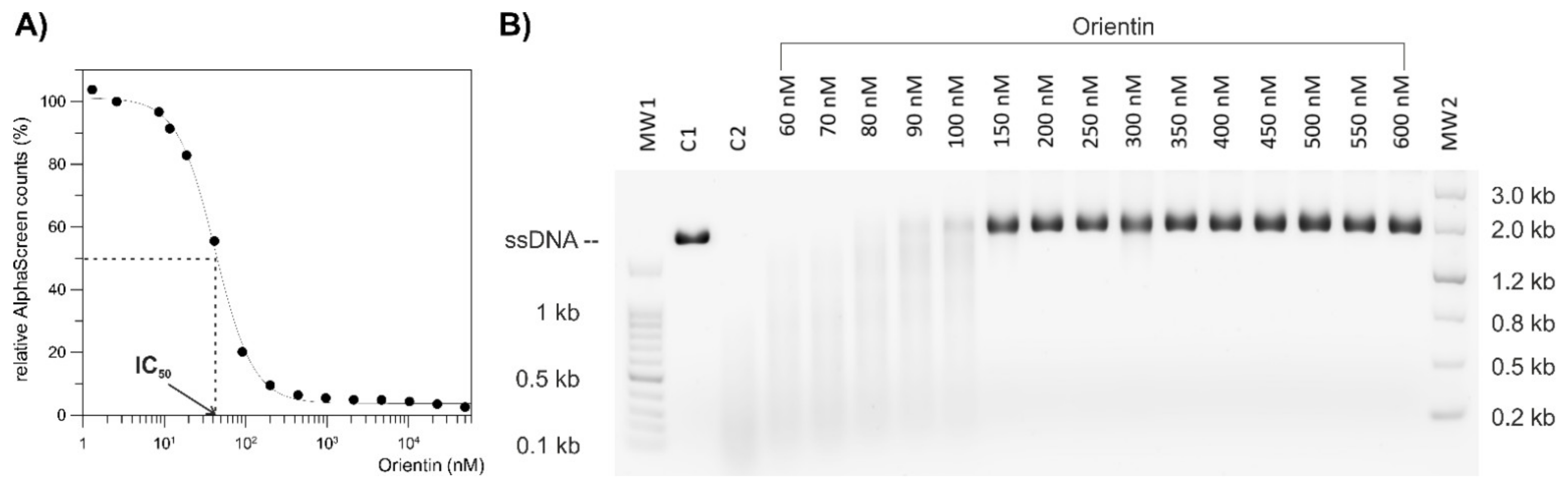
To compare the endonuclease activities of wild-type and I38T PA-Nter in the presence of selected compounds (baloxavir acid, luteolin, orientin), we used a gel-based endonuclease inhibitory assay. The single-stranded DNA substrate M13mp18 (New England Biolabs) was cleaved in vitro by either protein. Each reaction (10 µL) contained 1 μM protein (GST-PA-Nter wild type/GST-PA-Nter I38T mutant) in digestion buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween20, 1 mM MnCl2, 10 mM MgCl2, 1 mM 2-mercaptoethanol) and was incubated with various concentrations of inhibitors (BXA/LU2/OTN/21/30). Reactions were initiated by the addition of 0.2 μg of M13mp18 plasmid. Reactions were incubated at 37 °C for 5 h and stopped by adding 1 μL of 0.2 M EDTA. Finally, cleavage of DNA substrate was visualized by agarose electrophoresis using 1% agarose gel stained with GelRed.
3.4. Crystallization and Diffraction Data Collection
Hexagonal bifrustum crystals of empty wild-type and I38T PA-Nter subunits were obtained by the hanging-drop vapor diffusion method. Protein solution (12 mg/mL) was mixed with crystallization reservoir solution (12.5% w/v PEG 1000, 12.5% w/v PEG 3350, 0.1% M MOPS/HEPES-Na pH 7.5, 0.06 M magnesium chloride, 0.06 M calcium chloride) and PA-Nter seed in a 1:1:0.2 ratio. Crystals grew at 18 °C until they reached approximately 0.2 mm in diameter. Ligands (100 mM solution in DMSO) were soaked in for 15 min (the final DMSO concentration did not exceed 5%). Crystals were harvested, flash-cooled by plunging into liquid nitrogen and stored at −196 °C.
Diffraction qualities were tested at BESSY II and data were collected at −173 °C on a home diffractometer (MicroMax-007 HF microfocus equipped with a PILATUS 300 K detector, Rigaku). The crystal of wild-type PA-Nter soaked with orientin diffracted to a resolution of up to 1.87 Å, and the I38T PA-Nter/orientin crystal diffracted up to 2.15 Å. Diffraction data were processed, integrated, and reduced using XDS [
53] and scaled using XSCALE from the XDS suite [
54]. Both crystals belonged to the
P6
422 space group and contained one molecule per asymmetric unit, with a solvent content of 47.5% (wild-type PA-Nter), and 48.2% (I38T PA-Nter). We observed anomalous signals up to a resolution of 3.0 Å for wild-type and 3.5 Å for the I38T variant. Therefore, the data were processed with unmerged Friedel pairs. Detailed crystal parameters and data collection statistics are given in
Table S1.
3.5. Structure Determination and Analyses
Structures of wild-type and I38T PA-Nter were determined by molecular replacement with MOLREP [
55] from the CCP4 package [
56] using a previously reported structure of PA-Nter as a template (PDB entry 6YA5 [
37]). The final step of complex structure polishing was carried out by cycles of manual adjustments using Coot software [
57] followed by refinement in REFMAC 5.8.0103 [
58]. MolProbity [
59] was used to validate the quality of the final models. Refinement statistics are given in
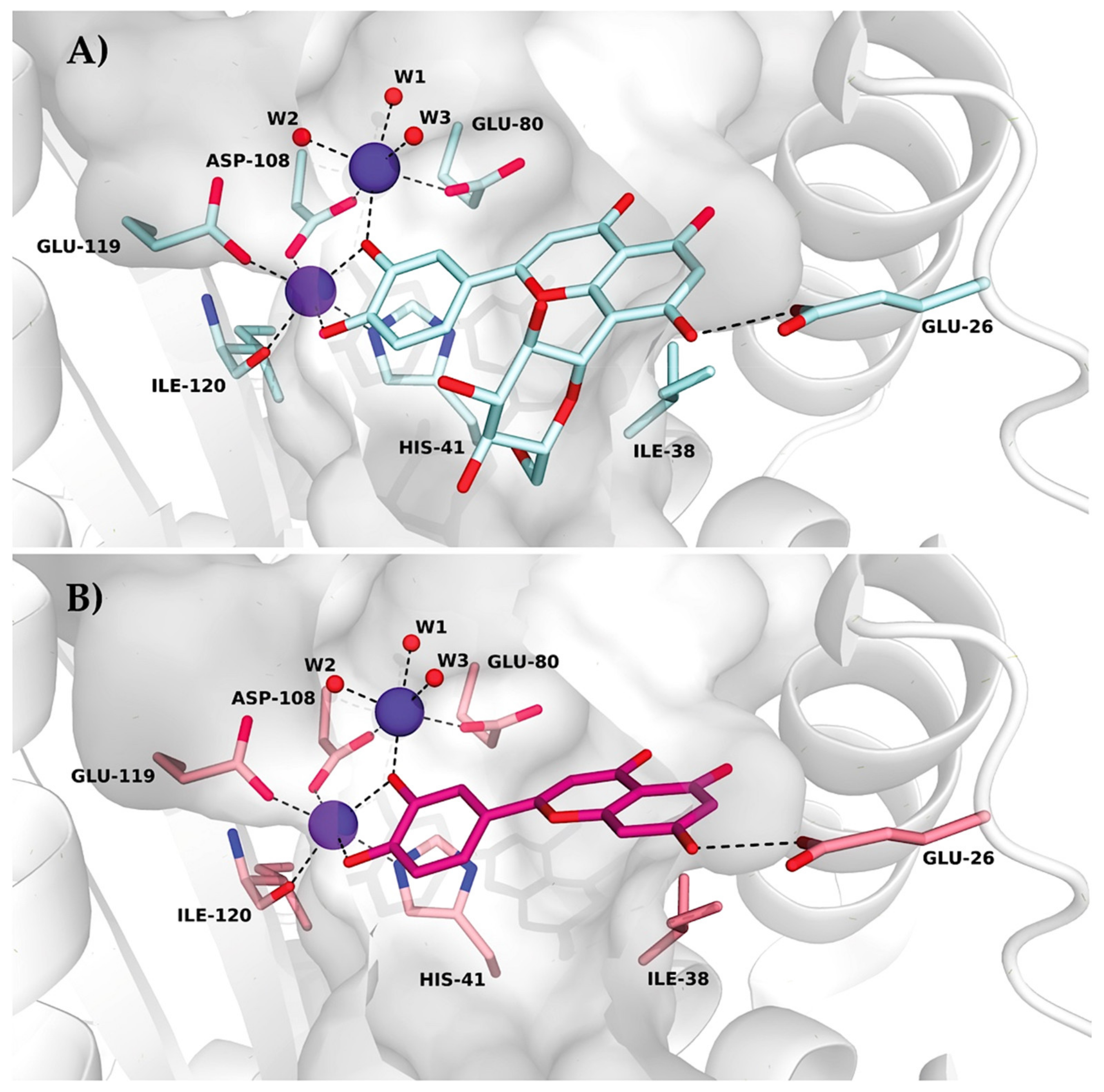
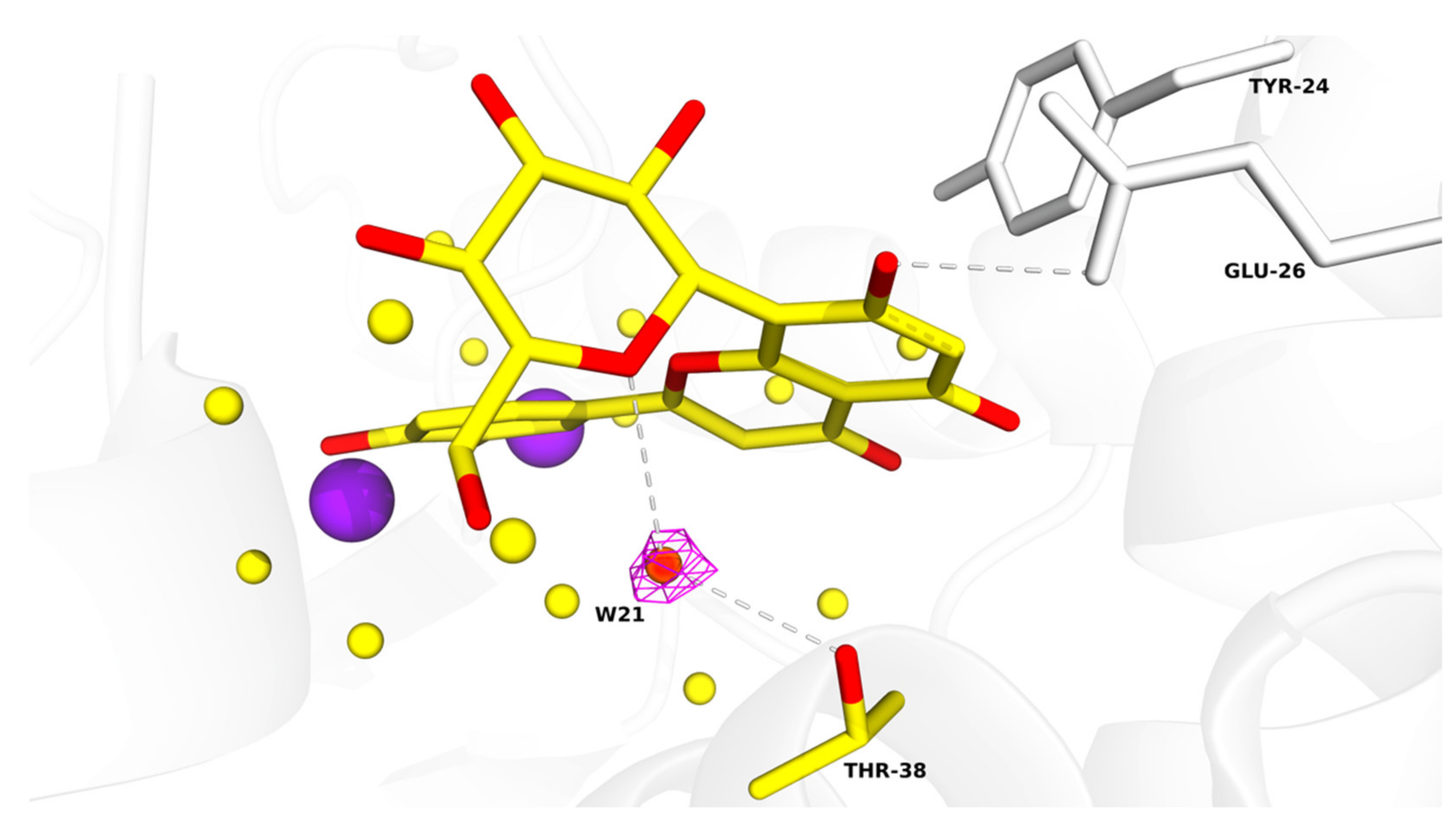
Table S1. All figures illustrating structural representations were prepared with PyMOL (The PyMOL Molecular Graphics System, Version 2.4.2 accessed on 10 March 2020; Schrödinger, LLC., New York, NY, USA). Atomic coordinates and experimental structure factors are deposited in the Protein Data Bank under codes 7NUG for wild-type PA-Nter in complex with orientin and 7NUH for I38T PA-Nter in complex with orientin.
3.6. Chemistry
Unless otherwise noted, all reactions were carried out under argon in oven-dried glassware. Solvents were distilled from drying agents as indicated and transferred under nitrogen: THF (Na/benzophenone), toluene (Na/benzophenone), MeCN (CaH2), and DCM (CaH2). Chromatography was performed using a Teledyne ISCO Combi Flash Rf+ flash chromatography system with RediSep Rf Gold Silica or RediSep Rf Gold Reversed-phase C18 columns. All starting materials were used as purchased (Sigma Aldrich, Alfa Aesar, TCI, Fluorochem, Combi-Blocks), unless otherwise indicated. Compounds luteolin and orientin were purchased from Sigma-Aldrich (product numbers L9283 and O9765). All inhibitors were purified using an ECOM TOY18DAD800 compact preparative system [flow rate 15 mL/min; gradient 0–60% MeCN/H2O (0.1% trifluoroacetic acid) over 60 min], with a ProntoSIL 120-10-C18 ace-EPS column, 10 μm, 20 × 250 mm. The purity of compounds and composition of the reaction mixtures were tested on a Waters UPLC-MS Acquity with QDa Mass Detector (flow rate 0.5 mL/min, gradient 0–100 % MeCN/H2O (0.1% formic acid) over 7 min) with an ACQUITY UPLC BEH C18 Column, 130 Å, 1.7 µm, 2.1 mm × 100 mm with a 2.1 mm × 5 mm pre-column. The final inhibitors were of at least 90% purity. 1H-NMR spectra were recorded on Bruker instruments at 400, 500 or 600 MHz; 13C-NMR spectra were recorded at 100, 126 or 150 MHz. Chemical shifts are provided in δ-scale; coupling constants J are given in Hz. ESI high resolution mass spectra were recorded using a Thermo Scientific LTQ Orbitrap XL (Termo Fisher Scientific, Waltham, Massachusetts, USA) controlled by MassLynx software.
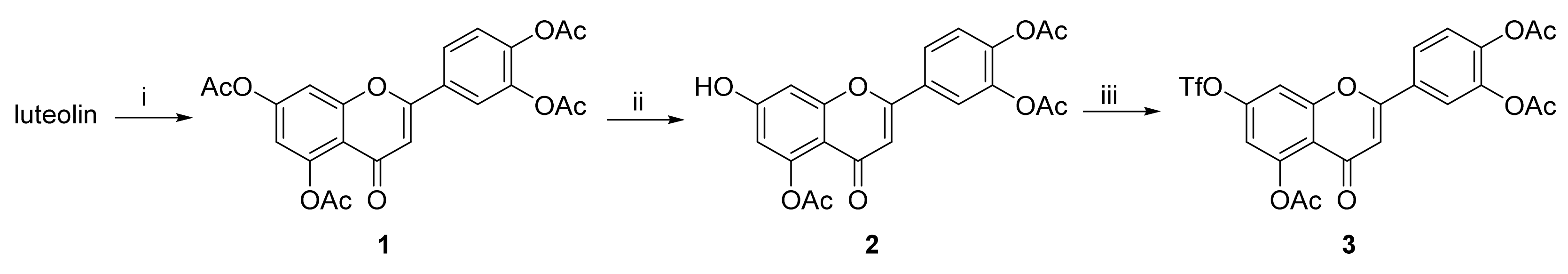
Compounds
1 and
2 were prepared according to literature procedures [
43]. Analytical data for these compounds were in agreement with published data.
3.7. 4-(5,7-Diacetoxy-4-oxo-4H-chromen-2-yl)-1,2-phenylene Diacetate (1)
Neat acetic anhydride (10 mL) was added to luteolin (1.43 g, 5.0 mmol), followed by the addition of pyridine (1.0 mL). The reaction mixture was heated to 145 °C for 3 h. The hot mixture was poured into ice and stirred for 30 min. The suspension was filtered off, and the solids were mixed with a boiling mixture of MeOH/CHCl3 (9:1, 15 mL). The product was isolated by filtration from the cool suspension to furnish the desired compound 1 (1.75 g, 77%). 1H NMR (400 MHz, CDCl3) δ = 7.74 (dd, J = 8.4, 2.2 Hz, 1H), 7.70 (d, J = 2.1 Hz, 1H), 7.38–7.32 (m, 2H), 6.85 (d, J = 2.2 Hz, 1H), 6.61 (s, 1H), 2.44 (s, 3H), 2.35 (s, 6H), 2.33 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ = 176.3, 169.5, 168.1, 167.9, 160.9, 157.7, 154.2, 150.4, 145.0, 142.8, 129.8, 124.6, 124.5, 121.8, 115.0, 114.0, 109.2, 109.1, 21.3, 21.2, 20.8, 20.8 ppm.
3.8. 4-(5-Acetoxy-7-hydroxy-4-oxo-4H-chromen-2-yl)-1,2-phenylene Diacetate (2)
4-(5,7-Diacetoxy-4-oxo-4H-chromen-2-yl)-1,2-phenylene diacetate (1) (1.70 g, 3.7 mmol, 1.00 eq.) was dissolved in THF (56 mL) and NMP (19 mL). Then, imidazole (88.5 mg, 1.3 mmol, 0.35 eq.) was added at 0 °C followed by addition of thiophenol (0.45 mL, 4.4 mmol, 1.18 eq.) via septum under a nitrogen atmosphere at 0 °C. The reaction mixture was stirred and slowly warmed to room temperature over 2 h until the starting material was fully consumed (TLC, UPLC-MS). Volatiles were evaporated and the oily residue was dissolved in EtOAc (150 mL). The organic phase was washed with 5% HCl (aq., 5 × 45 mL), and organic solvents were evaporated. The solids were mixed with EtOH, and the product was isolated by filtration to furnish the desired compound 2 (1.05 g, 68%). 1H NMR (400 MHz, DMSO-d6) δ = 11.17 (s, 1H), 8.08–7.95 (m, 2H), 7.49 (d, J = 9.0 Hz, 1H), 6.97 (d, J = 2.3 Hz, 1H), 6.80 (s, 1H), 6.60 (d, J = 2.3 Hz, 1H), 2.35 (s, 3H), 2.34 (s, 3H), 2.32 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 175.2, 168.9, 168.2, 168.0, 162.3, 159.4, 158.2, 150.1, 144.6, 142.5, 129.5, 124.8, 124.5, 121.7, 109.5, 108.8, 108.0, 101.0, 21.0, 20.4 (2C) ppm.
3.9. 4-(5-Acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene Diacetate (3)
A flask with 4-(5-acetoxy-7-hydroxy-4-oxo-4H-chromen-2-yl)-1,2-phenylene diacetate (2) (1.00 g, 2.4 mmol, 1.0 eq.) was purged with nitrogen. The solids were mixed with dry CH2Cl2 (12 mL) and the suspension was cooled to 0 °C. Pyridine (0.39 mL, 4.9 mmol, 2.0 eq.) was added dropwise. After 10 min, trifluoromethanesulfonic anhydride (0.53 mL, 3.2 mmol, 1.3 eq.) was added dropwise at 0 °C under a nitrogen atmosphere. The resulting red-colored reaction mixture was stirred for 3.5 h until the starting material was fully consumed (TLC, UPLC-MS). Then, the reaction mixture was diluted with additional CH2Cl2 (15 mL) and washed with sat. NH4Cl (aq., 1 × 10 mL), sat. CuSO4 (aq., 2 × 10 mL) and water (1 × 10 mL). The organic phase was dried over anhydrous MgSO4 and then evaporated under reduced pressure. The residue was purified by flash chromatography (SiO2, cyclohexane/EtOAc = 100:0 → 50:50) to afford the desired triflate 3 (533 mg, 41%). 1H NMR (400 MHz, CDCl3) δ = 7.74 (dd, J = 8.5, 2.3 Hz, 1H), 7.71 (d, J = 2.1 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 2.4 Hz, 1H), 6.65 (s, 1H), 2.45 (s, 3H), 2.35 (s, 3H), 2.33 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ = 175.6, 169.1, 168.1, 167.8, 161.3, 157.6, 151.6, 151.3, 145.3, 142.9, 129.2, 124.7, 124.6, 121.9, 120.4, 117.1, 113.6, 109.5, 109.4, 21.1, 20.8, 20.7 ppm. 19F NMR (376 MHz, CDCl3) δ = −72.4 ppm. HRMS (ESI) m/z calcd for C22H16F3O11S [M+H+]+ 545.0359, found 545.0356.
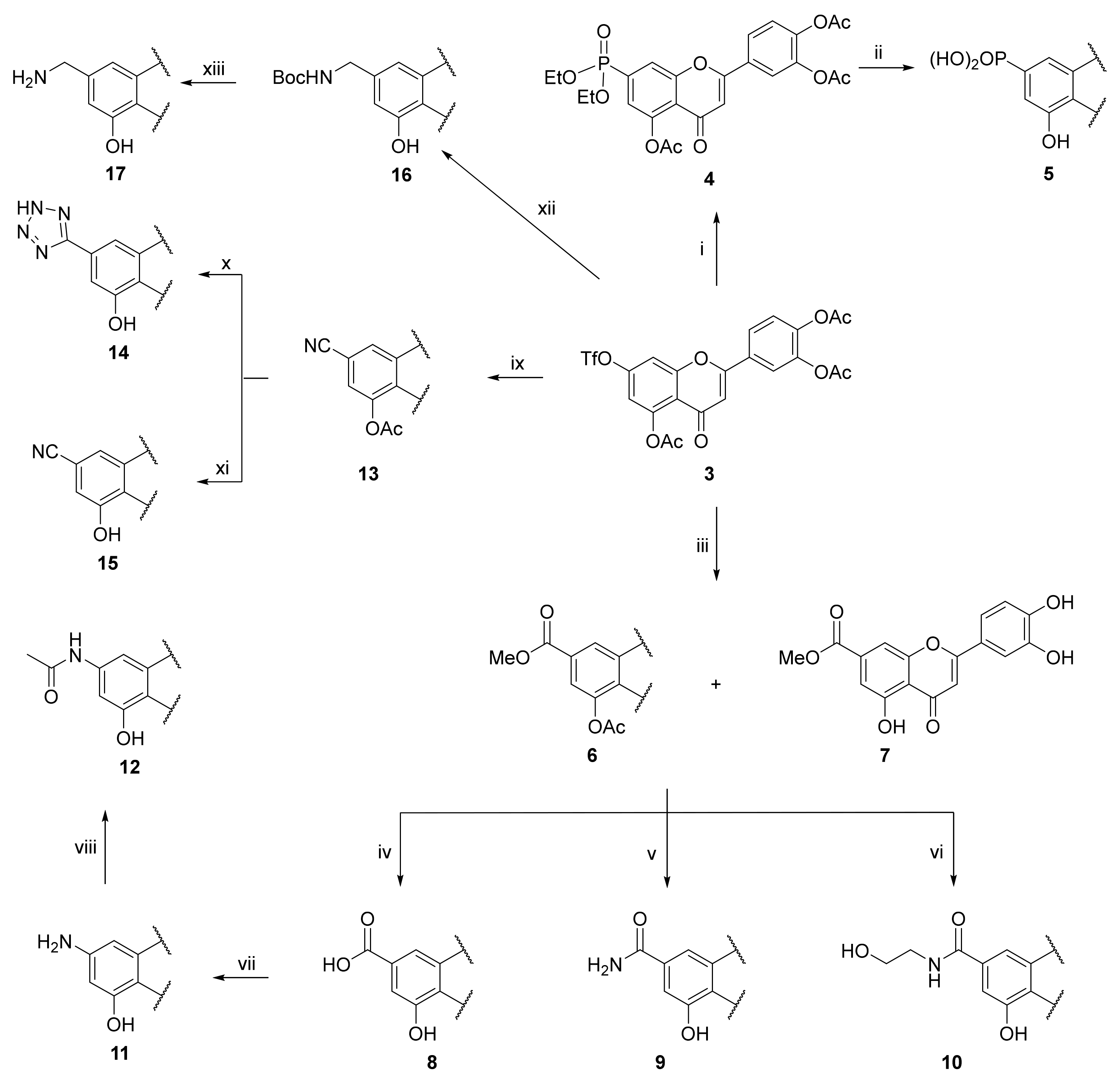
3.10. 4-(5-Acetoxy-7-(diethoxyphosphoryl)-4-oxo-4H-chromen-2-yl)-1,2-phenylene Diacetate (4)
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.0 eq.) and Pd(PPh3)4 (64 mg, 0.055 mmol, 0.3 eq.) was sealed, and the mixture was dissolved in anhydrous MeCN (1.8 mL) followed by an addition of diethylphosphite (30 μL, 0.221 mmol, 1.2 eq.) and DIPEA (42 μL, 0.240 mmol, 1.3 eq.) via septum. The mixture was degassed with a stream of argon for 15 min followed by heating to 70 °C for 3 h until the starting material was fully consumed (TLC, UPLC-MS). The reaction mixture was cooled to room temperature, filtered through Celite, and washed with EtOAc. Solvents were evaporated and the resulting mixture was purified by flash chromatography (SiO2, cyclohexane/EtOAc = 100:0 → 0:100) to afford the desired phosphonate 4 (76 mg, 78%). 1H NMR (400 MHz, CDCl3) δ = 7.93 (dd, J = 14.9, 1.3 Hz, 1H), 7.76–7.70 (m, 2H), 7.39–7.31 (m, 2H), 6.65 (s, 1H), 4.27–4.04 (m, 4H), 2.42 (s, 3H), 2.32 (s, 3H), 2.30 (s, 3H), 1.34 (td, J = 7.0, 0.6 Hz, 6H) ppm. 13C NMR (101 MHz, CDCl3) δ = 176.3, 169.5, 168.0, 167.8, 161.2, 156.8 (d, J = 23.5 Hz), 149.6, 149.4, 145.1, 142.8, 135.9, 134.0, 129.4, 124.6, 124.5, 121.8, 121.5 (d, J = 9.5 Hz), 120.3 (d, J = 10.3 Hz), 119.4 (d, J = 2.6 Hz), 109.2, 63.1, 63.0, 21.1, 20.7, 20.6, 16.4, 16.4 ppm. 31P NMR (162 MHz, CDCl3) δ = 16.58 ppm. HRMS (ESI) m/z calcd for C25H25O11PNa [M+Na+]+ 555.1026, found 555.1023.
3.11. (2-(3,4-Dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromen-7-yl)phosphonic Acid (5)
4-(5-Acetoxy-7-(diethoxyphosphoryl)-4-oxo-4H-chromen-2-yl)-1,2-phenylene diacetate (4) (312 mg, 0.58 mmol, 1.0 eq.) was dissolved in dry CH2Cl2 (18 mL). Then, bromo-trimethyl-silane (0.77 mL, 5.86 mmol, 10 eq.) was added dropwise under a nitrogen atmosphere. The reaction mixture was stirred for 31 h until the starting material was fully consumed (TLC, UPLC-MS). The solvent was evaporated and MeOH (3.1 mL) was added. The white suspension was allowed to stir for 1 h followed by evaporation of the solvent. THF (5.8 mL) was added, followed by an addition of 1 M KOH in MeOH (2.9 mL, 2.9 mmol, 5.0 eq). The reaction mixture was allowed to stir for 1 h. Solvent was evaporated, and the resulting mixture was purified by flash chromatography (SiO2-C18, H2O (0.1% TFA)/MeCN= 100:0 → 30:70) to afford the desired final phosphonic acid 5 (51 mg, 25%). 1H NMR (400 MHz, DMSO-d6) δ = 12.83 (s, 1H), 7.51 (dd, J = 8.3, 2.3 Hz, 1H), 7.48 (d, J = 2.3 Hz, 1H), 7.34 (dd, J = 14.0, 1.2 Hz, 1H), 6.97 (dd, J = 13.7, 1.2 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.87 (s, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 183.1, 165.7, 159.9 (d, J = 19.7 Hz), 155.8, 155.6, 150.7, 146.3, 143.6, 141.9, 121.6, 120.0, 116.5, 114.2, 112.5 (d, J = 9.9 Hz), 111.5 (d, J = 2.6 Hz), 109.6 (d, J = 9.9 Hz), 104.2 ppm. 31P NMR (162 MHz, DMSO) δ = 11.98 ppm. HRMS (ESI) m/z calcd for C15H10O8P [M−H+]– 349.0118, found 349.0118.
3.12. Palladium-Catalyzed Methoxycarbonylation of Triflate 3
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.0 eq.), 1,3-bis(diphenylphosphino)propane (7.50 mg, 0.018 mmol, 0.1 eq.), Mo(CO)6 (24 mg, 0.092 mmol, 0.5 eq.) and palladium acetate (4.00 mg, 0.018 mmol, 0.1 eq.) was sealed, and dry DMSO (1.1 mL) and dry MeOH (0.7 mL) were added via septum. The mixture was degassed with a stream of argon for 15 min followed by addition of triethylamine (57 μL, 0.405 mmol, 2.2 eq.). The reaction mixture was heated to 70 °C for 20 h until the starting material was fully consumed (UPLC-MS). The reaction mixture was cooled to room temperature, diluted with MeOH (5 mL), and filtered through a syringe filter. The solvents were evaporated, and the residue was purified by flash chromatography (SiO2-C18, H2O (0.1% TFA)/MeCN = 100:0 → 20:80) to afford the desired monoacetylated ester 6 (28 mg, 40%) and deacetylated ester 7 (29 mg, 48%).
3.13. Methyl 5-acetoxy-2-(3,4-dihydroxyphenyl)-4-oxo-4H-chromene-7-carboxylate (6)
1H NMR (401 MHz, DMSO-d6) δ 9.98 (s, 1H), 9.37 (s, 1H), 8.12 (t, J = 1.5 Hz, 1H), 7.56 – 7.52 (m, 1H), 7.50 – 7.43 (m, 2H), 6.90 (d, J = 8.2 Hz, 1H), 6.69 (s, 1H), 3.92 (s, 3H), 2.33 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ 175.2, 169.0, 164.2, 162.9, 156.4, 149.9, 148.8, 145.8, 133.9, 121.1, 119.3, 119.2, 118.8, 117.4, 116.0, 113.6, 106.2, 53.0, 20.9 ppm. HRMS (ESI) m/z calcd for C19H14O8Na [M+Na+]+ 393.0581, found 393.0580.
3.14. Methyl 2-(3,4-dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxylate (7)
1H NMR (401 MHz, DMSO-d6) δ 12.91 (s, 1H), 10.11 (s, 1H), 9.42 (s, 1H), 7.64 (d, J = 1.6 Hz, 1H), 7.55 (dd, J = 8.4, 2.3 Hz, 1H), 7.51 (d, J = 2.3 Hz, 1H), 7.22 (d, J = 1.6 Hz, 1H), 6.93–6.91 (m, 2H), 3.91 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ 182.9, 166.5, 165.3, 160.4, 156.0, 150.9, 146.3, 135.8, 121.4, 120.2, 116.5, 114.3, 113.0, 111.1, 108.7, 104.3, 53.3 ppm. HRMS (ESI) m/z calcd for C17H12O7 [M+H+]+ 329.0656, found 329.0655.
3.15. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxylic acid (8)
A solution of methyl 2-(3,4-dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxylate (7) (136 mg, 0.41 mmol, 1.0 eq.) in THF (8.4 mL) was heated to 45 °C under a nitrogen atmosphere, followed by addition of 2 M aq. LiOH (4.2 mL, 8.3 mmol, 20 eq.) via septum. The reaction mixture was stirred for 1 h until the starting material was fully consumed (UPLC-MS). The solvents were evaporated and the residue was purified by flash chromatography (SiO2-C18, H2O (0.1% TFA)/MeCN= 100:0 → 20:80) to afford the desired final carboxylic acid 8 (93 mg, 71 %). 1H NMR (400 MHz, DMSO-d6) δ = 12.87 (s, 1H), 10.09 (s, 1H), 9.43 (s, 1H), 7.61 (d, J = 1.3 Hz, 1H), 7.53 (dd, J = 8.4, 2.3 Hz, 1H), 7.49 (d, J = 2.3 Hz, 1H), 7.21 (d, J = 1.3 Hz, 1H), 6.91 (d, J = 9.0 Hz, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.6, 165.8, 165.6, 159.9, 155.5, 150.4, 145.9, 136.9, 121.1, 119.7, 116.1, 113.8, 112.3, 110.8, 108.2, 103.8 ppm. HRMS (ESI) m/z calcd for C16H10O7 [M−H+]– 313.0354, found 313.0351.
3.16. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxamide (9)
A tube with a mixture of methyl 5-acetoxy-2-(3,4-dihydroxyphenyl)-4-oxo-4H-chromene-7-carboxylate (6) (21.5 mg, 0.058 mmol, 1.0 eq.) and methyl 2-(3,4-dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxylate (7) (22 mg, 0.067 mmol, 1.15 eq.) was sealed, and 7 M ammonia in MeOH (10 mL) was added via septum. The reaction mixture was heated to 50 °C and stirred for 4 days until the starting materials were fully consumed (UPLC-MS). The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final amide 9 (8.0 mg, 21%). 1H NMR (600 MHz, DMSO-d6) δ = 12.82 (s, 1H), 10.07 (s, 1H), 9.45 (d, J = 2.5 Hz, 1H), 8.20 (s, 1H), 7.68 (s, 1H), 7.60 (d, J = 1.5 Hz, 1H), 7.51 (dd, J = 8.4, 2.3 Hz, 1H), 7.47 (d, J = 2.3 Hz, 1H), 7.25 (d, J = 1.3 Hz, 1H), 6.92 (d, J = 8.5 Hz, 1H), 6.89 (s, 1H) ppm. 13C NMR (151 MHz, DMSO-d6) δ = 182.6, 166.1, 165.5, 159.8, 155.5, 150.3, 145.9, 140.7, 121.2, 119.5, 116.1, 113.7, 111.3, 109.6, 106.6, 103.8 ppm. HRMS (ESI) m/z calcd for C16H11NO6Na [M+Na+]+ 336.0479, found 336.0479.
3.17. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-N-(2-hydroxyethyl)-4-oxo-4H-chromene-7-carboxamide (10)
Methyl 2-(3,4-dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxylate (7) (70 mg, 0.213 mmol, 1.0 eq.) was mixed with dry EtOH (2 mL). Potassium carbonate (100 mg, 0.723 mmol, 3.4 eq.) was added, followed by 2-aminoethanol (0.6 mL). The reaction mixture was heated to 50 °C and stirred for 48 h until the starting materials was fully consumed (UPLC-MS). The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final amide 10 (17 mg, 23%). 1H NMR (400 MHz, DMSO-d6) δ = 12.81 (s, 1H), 10.07 (s, 1H), 9.45 (s, 1H), 8.68 (t, J = 5.6 Hz, 1H), 7.59 (d, J = 1.5 Hz, 1H), 7.51 (dd, J = 8.3, 2.3 Hz, 1H), 7.48 (d, J = 2.3 Hz, 1H), 7.23 (d, J = 1.5 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.89 (s, 1H), 3.53 (t, J = 6.1 Hz, 2H), 3.35 (q, J = 5.2, 4.4 Hz, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.6, 165.4, 164.6, 159.8, 155.5, 150.3, 145.9, 140.9, 121.2, 119.5, 116.1, 113.7, 111.2, 109.3, 106.4, 103.7, 59.5, 42.4 ppm. HRMS (ESI) m/z calcd for C18H15NO7Na [M+Na+]+ 380.0741, found 380.0742.
3.18. 7-Amino-2-(3,4-dihydroxyphenyl)-5-hydroxy-4H-chromen-4-one (11)
2-(3,4-Dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carboxylic acid (8) (32 mg, 0.102 mmol, 1.0 eq.) was dissolved in dry DMF (1 mL) followed by dropwise addition of triethylamine (21 µL, 0.143 mmol, 1.4 eq.) and diphenylphosphoryl azide (31 µL, 0.143 mmol, 1.4 eq.) under a nitrogen atmosphere. The reaction mixture was heated to 100 °C and stirred for 16 h. Water (1 mL) was added and the reaction mixture was stirred for 3 h at 100 °C. The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final amine 11 (8.7 mg, 30%). 1H NMR (500 MHz, DMSO-d6) δ = 12.93 (s, 1H), 9.83 (s, 1H), 9.39 (s, 1H), δ = 7.36–7.33 (m, 2H), 6.87 (d, J = 8.1 Hz, 1H), 6.51 (s, 1H), 6.12 (d, J = 1.8 Hz, 1H), 5.92 (d, J = 2.0 Hz, 1H) ppm. 13C NMR (126 MHz, DMSO-d6) δ = 181.1, 163.2, 161.6, 158.1, 156.3, 149.7, 146.1, 122.3, 119.0, 116.5, 113.6, 103.0, 101.5, 96.3, 90.7 ppm. HRMS (ESI) m/z calcd for C15H12NO5 [M+H+]+ 286.0710, found 286.0710.
3.19. N-(2-(3,4-Dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromen-7-yl)acetamide (12)
7-Amino-2-(3,4-dihydroxyphenyl)-5-hydroxy-4H-chromen-4-one (11) (30 mg, 0.10 mmol, 1.0 eq.) was dissolved in dry DMF (0.5 mL), followed by addition of DIPEA (37 µL, 0.21 mmol, 2.0 eq.) and N-succinimidyl acetate (33 mg, 0.15 mmol, 1.5 eq.). The reaction mixture was allowed to stir at room temperature for 36 h until the starting material was fully consumed (UPLC-MS). The reaction mixture was directly purified by preparative HPLC to obtain the final product 12 (7.9 mg, 23%). 1H NMR (400 MHz, DMSO-d6) δ = 10.57 (s, 1H), 7.78 (dd, J = 8.6, 2.3 Hz, 1H), 7.73 (d, J = 2.2 Hz, 1H), 7.06 (d, J = 8.6 Hz, 1H), 6.67 (s, 1H), 6.17 (d, J = 1.8 Hz, 1H), 5.94 (d, J = 2.0 Hz, 1H), 2.30 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 180.6, 168.7, 161.7, 161.1, 157.6, 155.9, 152.5, 138.7, 125.2, 122.0, 121.5, 117.3, 103.2, 101.1, 95.9, 90.4, 20.6 ppm. HRMS (ESI) m/z calcd for C17H12NO6 [M−H+]– 326.0670, found 326.0671.
3.20. 4-(5-Acetoxy-7-cyano-4-oxo-4H-chromen-2-yl)-1,2-phenylene Diacetate (13)
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.00 eq.), zinc cyanide (12.0 mg, 0.101 mmol, 0.55 eq.), and XantPhos Pd G2 (2.00 mg, 0.002 mmol, 0.01 eq.) was sealed and purged with nitrogen. Dry DMF (0.9 mL) and DIPEA (15 μL, 0.084 mmol, 0.15 eq.) were added. The reaction mixture was heated to 85 °C and stirred for 16 h until the starting material was fully consumed (TLC, UPLC-MS). Then the reaction mixture was cooled to room temperature, and EtOAc (5 mL) was added. The reaction mixture was washed with water (2 × 10 mL), and the organic phase was dried over anhydrous MgSO4 and then evaporated under reduced pressure. The resulting mixture was purified by flash chromatography (SiO2, cyclohexane/EtOAc = 100:0 → 60:40) to afford the desired nitrile 13 (47 mg, 61 %). 1H NMR (400 MHz, CDCl3) δ = 7.79 (d, J = 1.5 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.73–7.71 (m, 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 1.5 Hz, 1H), 6.67 (s, 1H), 2.46 (s, 3H), 2.35 (s, 3H), 2.34 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ = 175.5, 169.2, 168.0, 167.7, 161.4, 156.8, 150.2, 145.3, 142.8, 128.9, 124.6, 124.5, 122.1, 121.8, 120.6, 120.2, 116.9, 116.2, 109.5, 21.0, 20.7, 20.6 ppm. HRMS (ESI) m/z calcd for C22H15NO8Na [M+Na+]+ 444.0690, found 444.0691.
3.21. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-7-(2H-tetrazol-5-yl)-4H-chromen-4-one (14)
A tube with 4-(5-acetoxy-7-cyano-4-oxo-4H-chromen-2-yl)-1,2-phenylene diacetate (13) (58 mg, 0.134 mmol, 1.0 eq) and NaN3 (36 mg, 0.562 mmol, 4.0 eq.) was sealed and the mixture was dissolved in dry DMF (0.5 mL). The mixture was degassed with a stream of argon for 10 min, followed by addition of glacial acetic acid (2 drops) via septum and the reaction mixture was heated to 130 °C for 16 h until the starting material was fully consumed (TLC, UPLC-MS). The reaction mixture was cooled to room temperature, and 4 M HCl was added until the pH reached 2. The solvents were evaporated, and the residue was purified by preparative HPLC to obtain the final tetrazole derivative 14 (18 mg, 38%). 1H NMR (400 MHz, DMSO-d6) δ = 13.02 (s, 1H), 10.09 (s, 1H), 9.45 (s, 1H), 7.77 (d, J = 1.5 Hz, 1H), 7.52 (dd, J = 8.4, 2.4 Hz, 1H), 7.49 (d, J = 2.3 Hz, 1H), 7.39 (d, J = 1.6 Hz, 1H), 6.98 –6.86 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.3, 165.4, 160.5, 156.1, 150.4, 145.9, 130.5, 121.0, 119.6, 116.1, 113.7, 111.2, 108.7, 105.8, 103.9 ppm. HRMS (ESI) m/z calcd for C16H11N4O5 [M+H+]+ 339.0724, found 339.0724.
3.22. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromene-7-carbonitrile (15)
4-(5-Acetoxy-7-cyano-4-oxo-4H-chromen-2-yl)-1,2-phenylene diacetate (13) (47 mg, 0.11 mmol, 1.0 eq.) was dissolved in THF (1.1 mL), followed by addition of 2 M aq. LiOH (0.33 mL, 0.66 mmol, 6.0 eq.). The reaction mixture was allowed to stir for 24 h at room temperature under a nitrogen atmosphere until the starting material was fully consumed (UPLC-MS). Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final nitrile 15 (25 mg, 77%). 1H NMR (400 MHz, DMSO-d6) δ = 13.12 (s, 1H), 10.15 (s, 1H), 9.46 (s, 1H), 7.72 (d, J = 1.3 Hz, 1H), 7.50 (dd, J = 8.3, 2.3 Hz, 1H), 7.46 (d, J = 2.3 Hz, 1H), 7.25 (d, J = 1.5 Hz, 1H), 6.94 (s, 1H), 6.91 (d, J = 8.3 Hz, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.3, 165.7, 160.3, 155.5, 150.6, 145.9, 120.8, 119.8, 117.4, 116.6, 116.0, 113.9, 112.8, 111.8, 104.2 ppm. HRMS (ESI) m/z calcd for C16H10NO5 [M+H+]+ 296.0554, found 296.0551.
3.23. Tert-Butyl ((2-(3,4-dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromen-7-yl)methyl)carbamate (16)
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.0 eq.), potassium (((tert-butoxy-carbonyl)amino)methyl)trifluoroborate (66 mg, 0.276 mmol, 1.5 eq.), K2CO3 (127 mg, 0.552 mmol, 3.0 eq.) and SPhos Pd G2 (15 mg, 0.018 mmol, 0.1 eq.) was sealed, and THF/water (25:1, 3.5 mL) was added via septum. The mixture was degassed with a stream of argon for 15 min, followed by heating to 100 °C for 2 h until the starting material was fully consumed (TLC, UPLC-MS). The reaction mixture was cooled to room temperature, diluted with EtOAc (2 mL), and filtered through a syringe filter. The solvents were evaporated and the residue was purified by flash chromatography (SiO2-C18, H2O/MeCN= 100:0 → 0:100) to afford the desired carbamate 16 (32 mg, 43%). 1H NMR (400 MHz, DMSO-d6) δ = 12.78 (s, 1H), 7.54 (t, J = 6.2 Hz, 1H), 7.47 (dd, J = 8.3, 2.3 Hz, 1H), 7.44 (d, J = 2.3 Hz, 1H), 6.98 (s, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.81 (s, 1H), 6.67 (d, J = 1.3 Hz, 1H), 4.21 (d, J = 6.1 Hz, 2H), 1.42 (s, 9H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 183.0, 165.2, 160.2, 156.2, 150.7, 149.8, 146.3, 121.6, 119.8, 116.5, 113.9, 109.5, 109.1, 105.5, 103.8, 78.6, 43.9, 28.7 ppm. HRMS (ESI) m/z calcd for C21H22NO7 [M+H+]+ 400.1391, found 400.1386.
3.24. 7-(Aminomethyl)-2-(3,4-dihydroxyphenyl)-5-hydroxy-4H-chromen-4-one (17)
Tert-butyl ((2-(3,4-dihydroxyphenyl)-5-hydroxy-4-oxo-4H-chromen-7-yl)methyl) carbamate (16) (25 mg, 0.063 mmol) was dissolved in THF/water (4:1, 1.5 mL), followed by addition of trifluoroacetic acid (0.6 mL). The reaction mixture was heated to 50 °C and allowed to stir for 48 h. The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final amine 17 (19 mg, 73%). 1H NMR (400 MHz, DMSO-d6) δ = 12.85 (s, 1H), 10.12 (s, 1H), 9.52 (s, 1H), 8.40 (s, 2H), 7.51–7.42 (m, 2H), 7.24 (d, J = 1.6 Hz, 1H), 6.97–6.90 (m, 2H), 6.87 (d, J = 1.8 Hz, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.5, 165.1, 159.9, 155.6, 150.3, 145.9, 142.1, 121.1, 119.3, 116.1, 113.6, 110.9, 109.5, 107.3, 103.7, 42.0 ppm. HRMS (ESI) m/z calcd for C16H14NO5 [M+H+]+ 300.0867, found 300.0870.
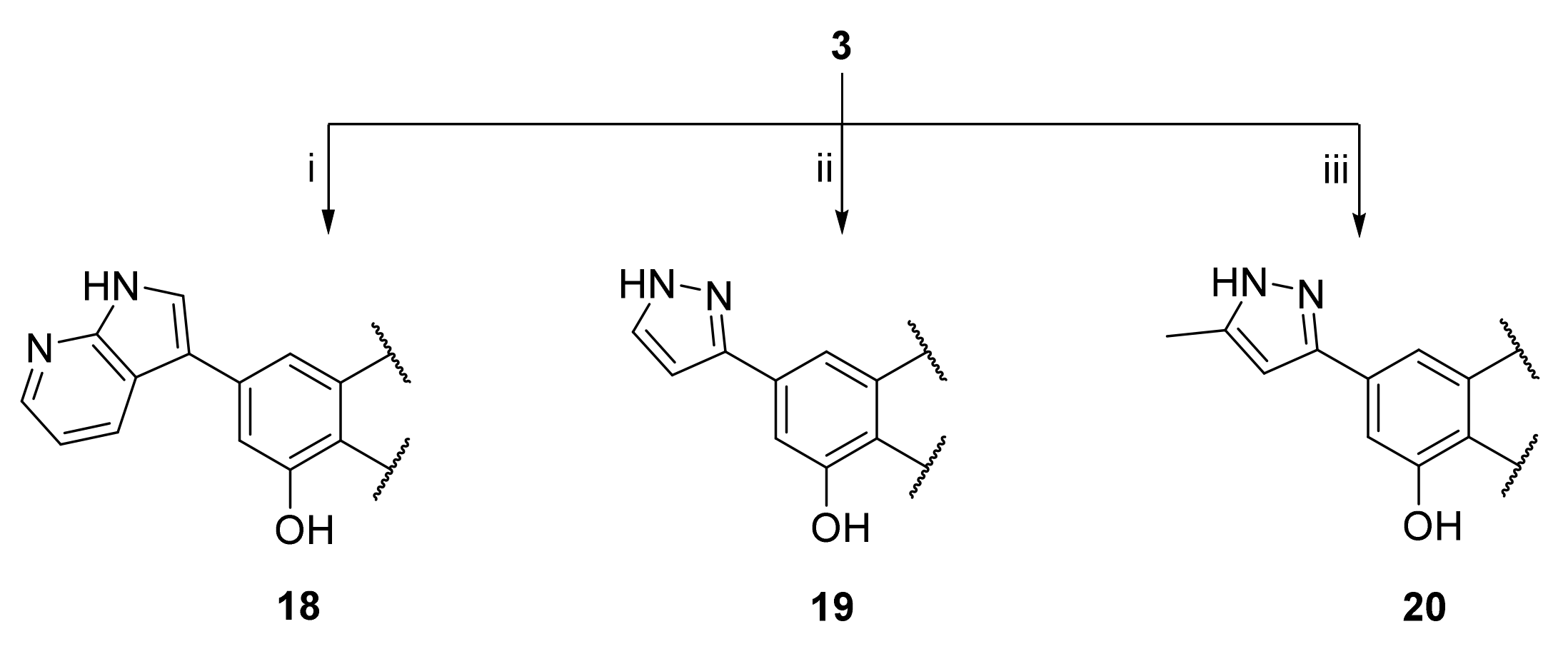
3.25. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-7-(1H-pyrrolo[2 ,3-B]pyridin-3-yl)-4H-chromen-4-one (18)
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.0 eq.), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrolo[2,3-b]pyridine (54 mg, 0.221 mmol, 1.2 eq.), Cs2CO3 (240 mg, 0.736 mmol, 4.0 eq.) and Pd(PPh3)4 (21 mg, 0.018 mmol, 0.1 eq.) was sealed, and dry THF (3.5 mL) was added via septum. The mixture was degassed with a stream of argon for 15 min. then heated to 100 °C for 16 h until the starting material was fully consumed (TLC, UPLC-MS). The reaction mixture was cooled to room temperature, diluted with MeOH (5 mL), and filtered through a syringe filter. The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 18 (6.6 mg, 10%). 1H NMR (400 MHz, DMSO-d6) δ = 12.87 (s, 1H), 12.27 (s, 1H), 10.02 (s, 1H), 9.42 (s, 1H), 8.49 (dd, J = 8.1, 1.6 Hz, 1H), 8.34 (dd, J = 4.6, 1.6 Hz, 1H), 8.25 (d, J = 2.8 Hz, 1H), 7.60 – 7.43 (m, 3H), 7.28–7.23 (m, 1H), 7.20 (d, J = 1.5 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.81 (s, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.6, 165.1, 160.6, 156.9, 150.4, 149.5, 146.3, 143.7, 143.2, 128.5, 127.3, 122.0, 119.8, 117.6, 117.2, 116.4, 114.2, 113.3, 108.3, 108.2, 104.4, 103.8 ppm. HRMS (ESI) m/z calcd for C22H13N2O5 [M−H+]– 385.0830, found 385.0827.
3.26. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-7-(1H-pyrazol-3-yl)-4H-chromen-4-one (19)
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.0 eq.), (1H-pyrazol-3-yl)boronic acid (31 mg, 0.276 mmol, 1.5 eq.), K2CO3 (127 mg, 0.920 mmol, 5.0 eq.), and SPhos Pd G2 (15 mg, 0.018 mmol, 0.1 eq.) was sealed, and dry THF (3.5 mL) was added via septum. The mixture was degassed with a stream of argon for 15 min, then heated to 100 °C for 16 h until the starting material was fully consumed (TLC, UPLC-MS). The reaction mixture was cooled to room temperature, diluted with MeOH (5 mL), and filtered through a syringe filter. The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 19 (15 mg, 25%). 1H NMR (400 MHz, DMSO-d6) δ = 12.84 (s, 1H), 7.83 (d, J = 2.3 Hz, 1H), 7.59 (d, J = 1.5 Hz, 1H), 7.52–7.46 (m, 2H), 7.27 (d, J = 1.5 Hz, 1H), 6.96 (d, J = 2.3 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.81 (s, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.4, 164.9, 160.1, 157.3, 156.3, 150.0, 147.3, 145.8, 140.1, 132.2, 121.4, 119.3, 116.0, 113.7, 109.0, 107.2, 103.6, 103.5 ppm. HRMS (ESI) m/z calcd for C18H13N2O5 [M+H+]+ 337.0819, found 337.0819.
3.27. 2-(3,4-Dihydroxyphenyl)-5-hydroxy-7-(5-methyl-1H-pyrazol-3-yl)-4H-chromen-4-one (20)
A tube with 4-(5-acetoxy-4-oxo-7-(((trifluoromethyl)sulfonyl)oxy)-4H-chromen-2-yl)-1,2-phenylene diacetate (3) (100 mg, 0.184 mmol, 1.0 eq.), (5-methyl-1H-pyrazol-3-yl)boronic acid (35 mg, 0.276 mmol, 1.5 eq.), K2CO3 (127 mg, 0.920 mmol, 5 eq.), and SPhos Pd G2 (15 mg, 0.018 mmol, 0.1 eq.) was sealed, and THF/water (25:1, 3.5 mL) was added via septum. The mixture was degassed with a stream of argon for 15 min, then heated to 100 °C for 16 h until the starting material was fully consumed (TLC, UPLC-MS). The reaction mixture was cooled to room temperature, diluted with MeOH (5 mL), and filtered through a syringe filter. Then solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 20 (12 mg, 19%). 1H NMR (400 MHz, DMSO-d6) δ = 7.51 (d, J = 1.5 Hz, 1H), 7.51–7.46 (m, 2H), 7.19 (d, J = 1.3 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.80 (s, 1H), 6.66 (d, J = 1.0 Hz, 1H), 2.28 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.3, 164.8, 160.1, 158.4, 158.1, 156.2, 150.0, 145.8, 140.3, 121.4, 119.3, 116.0, 113.7, 108.9, 107.0, 103.5, 103.3, 102.7, 10.9 ppm. HRMS (ESI) m/z calcd for C19H15N2O5 [M+H+]+ 351.0976, found 351.0975.
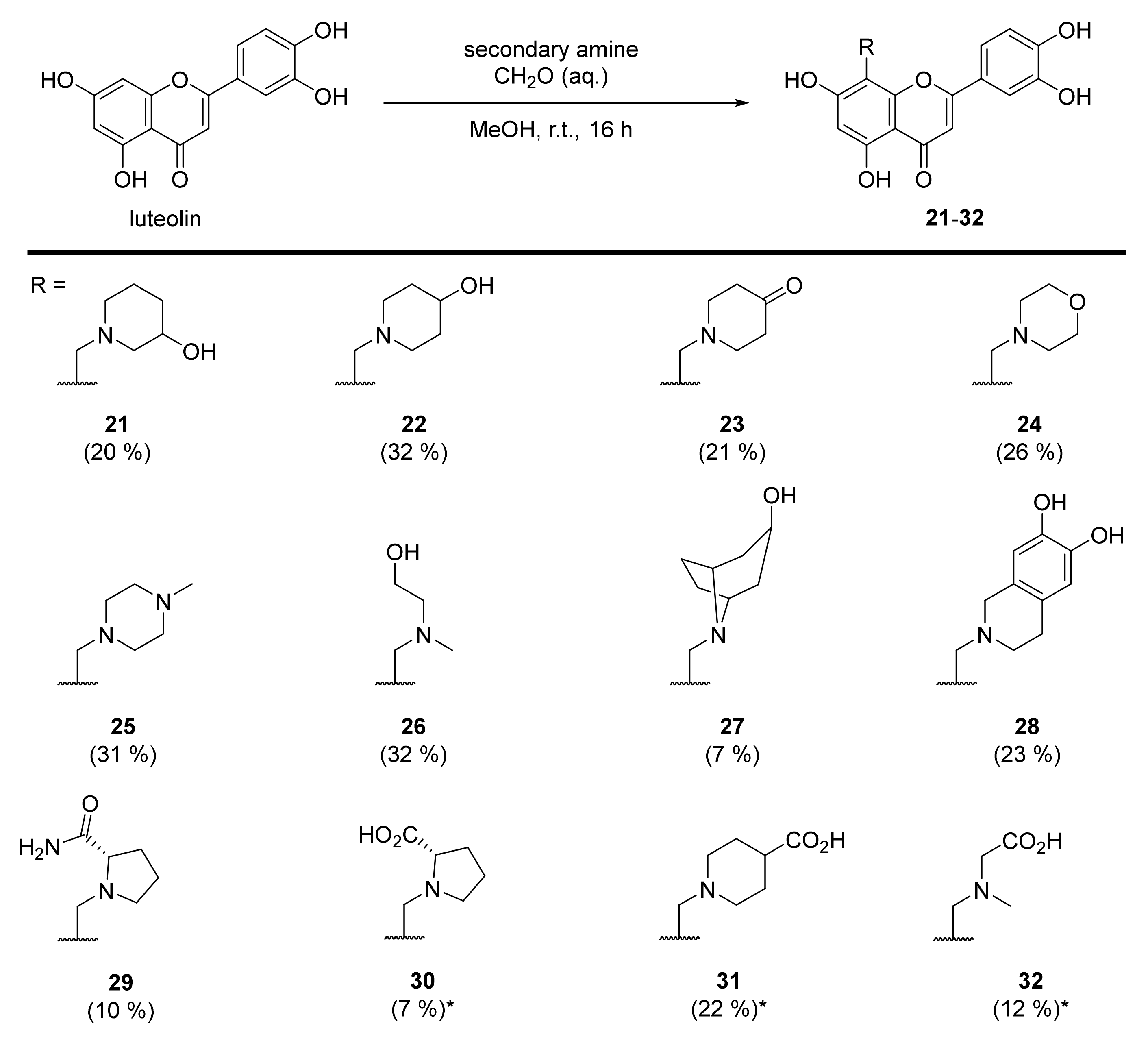
3.28. 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-8-((3-hydroxypiperidin-1-yl)methyl)-4H-chromen-4-one (21)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of 3-hydroxypiperidine (35 mg, 0.35 mmol, 1.0 eq.) and a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 21 (27 mg, 20%). 1H NMR (500 MHz, DMSO-d6, 330 K) δ = 13.27 (s, 1H), 9.89 (s, 1H), 9.35 (s, 1H), 7.47 (dd, J = 8.2, 2.3 Hz, 1H), 7.45 (d, J = 2.3 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 0.6 Hz, 1H), 6.39 (s, 1H), 5.33 (s, 1H), 4.63–4.29 (m, 2H), 4.01–2.80 (m, 5H), 2.11–1.31 (m, 4H) ppm. 13C NMR (126 MHz, DMSO-d6, 330 K) δ = 181.7, 164.1, 163.7, 162.5, 156.5, 150.0, 145.9, 121.5, 119.3, 116.2, 113.7, 103.9, 103.2, 98.4, 95.5, 57.3, 51.9, 49.1 ppm. HRMS (ESI) m/z calcd for C21H20NO7 [M−H+]– 398.1245, found 398.1244.
3.29. 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-8-((4-hydroxypiperidin-1-yl)methyl)-4H-chromen-4-one (22)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of 4-hydroxypiperidine (35 mg, 0.35 mmol, 1.0 eq.) and a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 22 (58 mg, 32%). 1H NMR (400 MHz, DMSO-d6) δ = 13.32 (s, 1H), 12.17 (br s, 1H), 10.12 (br s, 1H), 9.38 (br s, 1H), 7.56–7.44 (m, 2H), 6.95 (d, J = 9.0 Hz, 1H), 6.79 (s, 1H), 6.43 (s, 2H), 4.43 (br s, 2H), 3.91 (br s, 1H), 3.39 – 2.97 (m, 4H), 1.89–1.48 (m, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.2, 164.5, 164.2, 162.8, 156.8, 150.4, 146.3, 121.8, 119.7, 116.6, 113.8, 104.2, 103.5, 98.8, 96.1, 59.9, 51.1, 47.9, 31.7 ppm. HRMS (ESI) m/z calcd for C21H22NO7 [M+H+]+ 400.1391, found 400.1387.
3.30. 1-((2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl)methyl)piperidin-4-one (23)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of 4-piperidone monohydrate hydrochloride (54 mg, 0.35 mmol, 1.0 eq.), 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.), and triethylamine (49 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 23 as a 3:1 mixture of C-8 and C-6 regioisomers (38 mg, 21%). Spectral data includes the C-8 regioisomer only. 1H NMR (400 MHz, DMSO-d6) δ = 13.09 (s, 1H), 7.49–7.45 m, 2H), 6.93–6.91 (m, 1H), 6.73 (s, 1H), 6.29 (s, 1H), 3.97 (s, 2H), 2.93 (t, J = 5.8 Hz, 4H), 2.41 (t, J = 6.1 Hz, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 208.1, 183.3, 164.2, 164.0, 161.0, 155.8, 150.2, 146.2, 122.2, 119.5, 116.5, 113.9, 104.1, 103.1, 98.9, 94.1, 52.5, 49.4, 40.8 ppm. HRMS (ESI) m/z calcd for C21H18NO7 [M−H+]– 396.1089, found 396.1085.
3.31. 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-8-(morpholinomethyl)-4H-chromen-4-one (24)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of morpholine (30 µL, 0.35 mmol, 1.0 eq.) and a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 24 (46 mg, 26%). 1H NMR (400 MHz, DMSO-d6, 330 K) δ = 13.33 (s, 1H), 12.13 (br s, 1H), 10.07 (br s, 1H), 9.59 (br s, 1H), 7.51 (dd, J = 8.3, 2.3 Hz, 1H), 7.48 (d, J = 2.3 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.79 (s, 1H), 6.42 (s, 1H), 4.47 (s, 2H), 4.01–3.20 (m, 8H) ppm. 13C NMR (101 MHz, DMSO-d6, 330 K) δ = 181.8, 164.1, 163.9, 162.5, 156.4, 150.0, 145.9, 121.3, 119.4, 116.2, 113.6, 103.7, 103.0, 98.4, 95.2, 63.2, 51.5, 48.9 ppm. HRMS (ESI) m/z calcd for C20H18NO7 [M−H+]— 384.1089, found 384.1087.
3.32. 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-8-((4-methylpiperazin-1-yl)methyl)-4H-chromen-4-one (25)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of N-methyl-piperazine (40 µL, 0.35 mmol, 1.0 eq.) and a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 25 (44 mg, 31%). 1H NMR (500 MHz, DMSO-d6, 330 K) δ = 13.10 (br s, 1H), 7.46–7.41 (m, 2H), 6.93 (d, J = 8.7 Hz, 1H), 6.68 (s, 1H), 6.34 (s, 1H), 4.01 (br s, 2H), 3.34–2.87 (m, 8H), 2.75 (s, 3H) ppm. 13C NMR (126 MHz, DMSO-d6) δ = 181.9, 164.0, 163.3, 161.2, 155.9, 149.9, 145.9, 121.7, 119.1, 116.2, 113.6, 103.8, 102.9, 98.4, 51.9, 49.1, 48.4, 42.3 ppm. HRMS (ESI) m/z calcd for C21H21N2O6 [M−H+]– 397.1405, found 397.1403.
3.33. 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-8-(((2-hydroxyethyl)(methyl)amino)methyl)-4H-chromen-4-one (26)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of N-methylethanolamine (28 µL, 0.35 mmol, 1.0 eq.) and a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 26 (42 mg, 32%). 1H NMR (400 MHz, DMSO-d6) δ = 13.96 (s, 1H), 12.21 (br s, 1H), 10.08 (br s, 1H), 9.54 (br s, 1H), 7.47 – 7.41 (m, 2H), 6.91 (d, J = 9.0 Hz, 1H), 6.76 (s, 1H), 6.65 (s, 1H), 5.34 (br s, 1H), 4.27 (br s, 2H), 3.80 (t, J = 5.4 Hz, 2H), 3.25 (br s, 2H), 2.75 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 181.8, 164.4, 163.5, 161.3, 157.6, 150.1, 145.9, 121.2, 119.2, 116.1, 113.5, 103.3, 102.9, 100.3, 93.5, 57.6, 55.4, 47.7, 40.5 ppm. HRMS (ESI) m/z calcd for C19H20NO7 [M+H+]+ 374.1234, found 374.1235.
3.34. 2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-8-((3-hydroxy-8-azabicyclo[3.2.1]octan-8-yl)methyl)-4H-chromen-4-one (27)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by an addition of nortropine hydrochloride (58 mg, 0.35 mmol, 1.0 eq.), a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.), and triethylamine (49 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 27 (10 mg, 7%). 1H NMR (500 MHz, DMSO-d6) δ = 13.27 (s, 1H), 11.94 (br s, 1H), 10.09 (br s, 1H), 9.51 (br s, 1H), 8.83 (br s, 1H), 7.48 (dd, J = 8.3, 2.4 Hz, 1H), 7.43 (d, J = 2.4 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.81 (s, 1H), 6.38 (s, 1H), 4.95 (br s, 1H), 4.27 (s, 2H), 4.17–3.66 (m, 4H), 2.47–2.35 (m, 4H), 2.18 (br d, J = 15.4 Hz, 2H), 1.88 (br d, J = 14.3 Hz, 2H) ppm. 13C NMR (126 MHz, DMSO-d6) δ = 181.9, 164.3, 163.7, 162.5, 156.6, 150.2, 146.1, 121.7, 119.5, 116.3, 113.8, 104.0, 103.4, 98.5, 96.7, 62.1, 60.6, 44.9, 37.2 (2C), 24.4 (2C) ppm. HRMS (ESI) m/z calcd for C23H22NO7 [M−H+]– 424.1402, found 424.1400.
3.35. 8-((6,7-Dihydroxy-3,4-dihydroisoquinolin-2(1H)-yl)methyl)-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one (28)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in MeOH (5 mL), followed by addition of 6,7-dihydroxy-1,2,3,4-tetrahydroisoquinolin-2-ium chloride (71 mg, 0.35 mmol, 1.0 eq.), a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.), and triethylamine (49 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 28 (46 mg, 23 %). 1H NMR (400 MHz, DMSO-d6) δ = 13.29 (s, 1H), 12.07 (br s, 1H), 10.07 (br s, 1H), 9.62 (br s, 1H), 9.12 (br s, 1H), 9.06 (s, 1H), 7.52–7.46 (m, 1H), 7.45 (d, J = 2.3 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.79 (s, 1H), 6.57 (s, 1H), 6.53 (s, 1H), 6.41 (s, 1H), 4.54 (s, 2H), 4.36 (s, 2H), 3.58 (br s, 2H), 2.91 (t, J = 6.9 Hz, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 182.2, 164.4, 164.2, 162.8, 156.8, 150.4, 146.3, 145.9, 144.9, 121.8 (2C), 119.7, 118.9, 116.6, 115.3, 114.0, 113.5, 104.2, 103.4, 98.7, 96.1, 53.0, 49.8, 48.2, 24.8 ppm. HRMS (ESI) m/z calcd for C25H20NO8 [M−H+]– 462.1194, found 462.1193.
3.36. (S)-1-((2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl)methyl)pyrro-lidine-2-carboxamide (29)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in EtOH (5 mL), followed by an addition of L-prolinamide hydrochloride (71 mg, 0.35 mmol, 1.0 eq.), a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.), and triethylamine (49 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 29 (19 mg, 10%). 1H NMR (400 MHz, DMSO-d6) δ = 13.31 (s, 1H), 11.85 (br s, 1H), 10.08 (br s, 1H), 9.52 (br s, 1H), 9.38 (br s, 1H), 7.96 (s, 1H), 7.69 (s, 1H), 7.55 (dd, J = 8.4, 2.3 Hz, 1H), 7.47 (d, J = 2.3 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 6.77 (s, 1H), 6.34 (s, 1H), 4.54 (d, Jgem = 13.7 Hz, 1H), 4.48 (d, Jgem = 13.7 Hz, 1H), 4.16 (br d, J = 9.3 Hz, 1H), 3.56 (br s, 2H), 2.47–2.35 (m, 1H), 2.04–1.93 (m, 1H), 1.89–1.77 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 181.8, 169.3, 164.1, 163.6, 162.4, 156.4, 150.0, 145.8, 121.3, 119.6, 116.0, 113.9, 103.7, 103.0, 98.3, 96.0, 65.9, 53.9, 45.5, 29.2, 22.3 ppm. HRMS (ESI) m/z calcd for C21H21N2O7 [M+H+]+ 413.1343, found 413.1344.
3.37. ((2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl)methyl)-L-proline (30)
Luteolin (100 mg, 0.35 mmol, 1.0 eq.) was dissolved in EtOH (5 mL), followed by addition of L-proline tert-butyl ester hydrochloride (72 mg, 0.35 mmol, 1.0 eq.) and a 35% formalin solution (27 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was stirred at room temperature for 16 h. The solvent was evaporated under reduced pressure and the residue was purified by preparative HPLC to obtain the monosubstituted luteolin C-8 derivative as tert-butyl ester (UPLC-MS: m/z = 470). The monosubstituted luteolin C-8 derivative was used in the next step without further characterization. The tert-butyl ester was dissolved in a mixture of TFA/DCM (1:1, 1 mL) with 1,3-dimethoxybenzene (25 µL, 0.19 mmol, 0.54 eq.) as a scavenger. The reaction mixture was allowed to stir at room temperature for 16 h. The reaction mixture was concentrated using a stream of nitrogen, and the product was precipitated with tert-butyl methyl ether. The precipitate was collected and purified by preparative HPLC to obtain the product 30 (9.5 mg, 7% – over two steps). 1H NMR (500 MHz, DMSO-d6) δ = 13.25 (s, 1H), 9.91 (br s, 1H), 7.68 (d, J = 2.3 Hz, 1H), 7.48 (dd, J = 8.4, 2.3 Hz, 1H), 6.88 (d, J = 8.2 Hz, 1H), 6.77 (s, 1H), 6.31 (s, 1H), 4.50 (d, Jgem = 13.7 Hz, 1H), 4.39 (d, Jgem = 13.7 Hz, 1H), 4.03 (m, 1H), 3.39 (m, 1H), 3.13–2.98 (m, 1H), 2.39–2.31 (m, 1H), 1.96 (m, 2H), 1.79 (m, 1H) ppm. 13C NMR (126 MHz, DMSO-d6) δ = 181.9, 170.8, 164.0, 163.7, 161.9, 156.0, 149.8, 145.9, 121.2, 119.0, 116.1, 114.3, 103.7, 102.7, 98.3, 97.4, 67.0, 53.6, 46.1, 28.4, 22.5. HRMS (ESI) m/z calcd for C21H18NO8 [M−H+]– 412.1035, found 412.1037.
3.38. 1-((2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl)methyl)piperidine-4-carboxylic acid (31)
Luteolin (143 mg, 0.50 mmol, 1.0 eq.) was dissolved in MeOH (7 mL), followed by addition of ethyl 4-piperidinecarboxylate (77 µL, 0.50 mmol, 1.0 eq.) and a 35% formalin solution (38 µL, 0.35 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography (SiO2-C18, H2O (0.1% TFA)/MeCN= 100:0 → 20:80) to obtain the monosubstituted luteolin C-8 derivative as an ethyl ester (UPLC-MS: m/z = 456). The monosubstituted luteolin C-8 derivative was used in the next step without further characterization. The ethyl ester was dissolved in THF (2 mL) and 5% aq. HCl (2 mL) was added. The reaction mixture was allowed to stir at 70 °C for 7 h until the starting material was fully consumed (UPLC-MS). Then, solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 31 (60 mg, 22% – over 2 steps). 1H NMR (400 MHz, DMSO-d6) δ = 13.31 (s, 1H), 12.07 (br s, 1H), 10.09 (br s, 1H), 9.56 (br s, 1H), 7.49 (dd, J = 8.3, 2.3 Hz, 1H), 7.46 (d, J = 2.2 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.79 (s, 1H), 6.40 (s, 1H), 4.41 (s, 2H), 3.73–3.37 (m, 4H), 3.28–3.08 (m, 1H), 2.13–1.69 (m, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 181.8, 174.7, 164.1, 163.8, 162.5, 156.4, 150.0, 145.9, 121.3, 119.3, 116.2, 113.6, 103.8, 103.1, 98.4, 95.6, 51.5 (2C), 48.7, 37.6, 25.4 (2C) ppm. HRMS (ESI) m/z calcd for C22H20NO8 [M−H+]– 426.1194, found 426.1190.
3.39. N-((2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl)methyl)-N-methylglycine (32)
Luteolin (143 mg, 0.50 mmol, 1.0 eq.) was dissolved in MeOH (7 mL), followed by addition of sarcosine methyl ester hydrochloride (70 mg, 0.50 mmol, 1.0 eq.), a 35% formalin solution (38 µL, 0.35 mmol, 1.0 eq.), and triethylamine (70 µL, 0.50 mmol, 1.0 eq.). The reaction mixture was allowed to stir at room temperature for 16 h. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography (SiO2-C18, H2O (0.1% TFA)/MeCN= 100:0 → 40:60) to obtain the monosubstituted luteolin C-8 derivative as a methyl ester (UPLC-MS: m/z = 402). The monosubstituted luteolin C-8 derivative was used in the next step without further characterization. The methyl ester was dissolved in THF (2 mL) and 5% aq. HCl (2 mL) was added. The reaction mixture was allowed to stir at 70 °C for 3 days until the starting material was fully consumed (UPLC-MS). The solvents were evaporated and the residue was purified by preparative HPLC to obtain the final product 32 (29 mg, 12% – over 2 steps). 1H NMR (400 MHz, DMSO-d6) δ = 13.30 (s, 1H), 10.09 (br s, 1H), 9.54 (br s, 1H), 7.52 (dd, J = 8.3, 2.3 Hz, 1H), 7.49 (d, J = 2.3 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.76 (s, 1H), 6.35 (s, 1H), 4.49 (s, 2H), 4.00 (s, 2H), 2.75 (s, 3H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 181.8, 168.1, 164.0 (2C), 162.3, 156.4, 149.9, 145.9, 121.3, 119.3, 116.0, 113.8, 103.8, 103.0, 98.4, 96.1, 55.5, 48.2, 40.9 ppm. HRMS (ESI) m/z calcd for C19H18NO8 [M+H+]+ 388.1027, found 388.1026.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























