
NEUROD1 Is Required for the Early α and β Endocrine Differentiation in the Pancreas

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
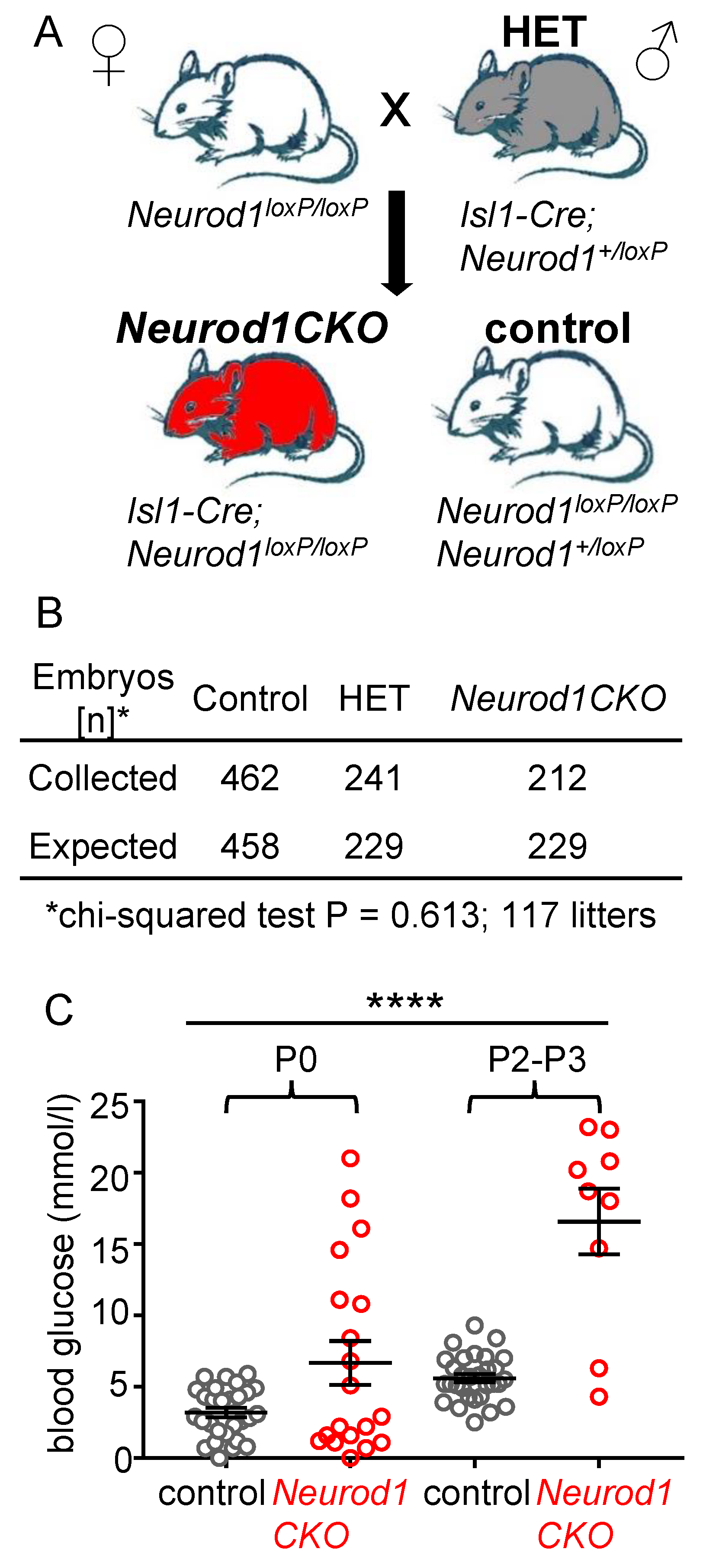
2.1. Conditional Deletion of Neurod1 Results in Neonatal Diabetes
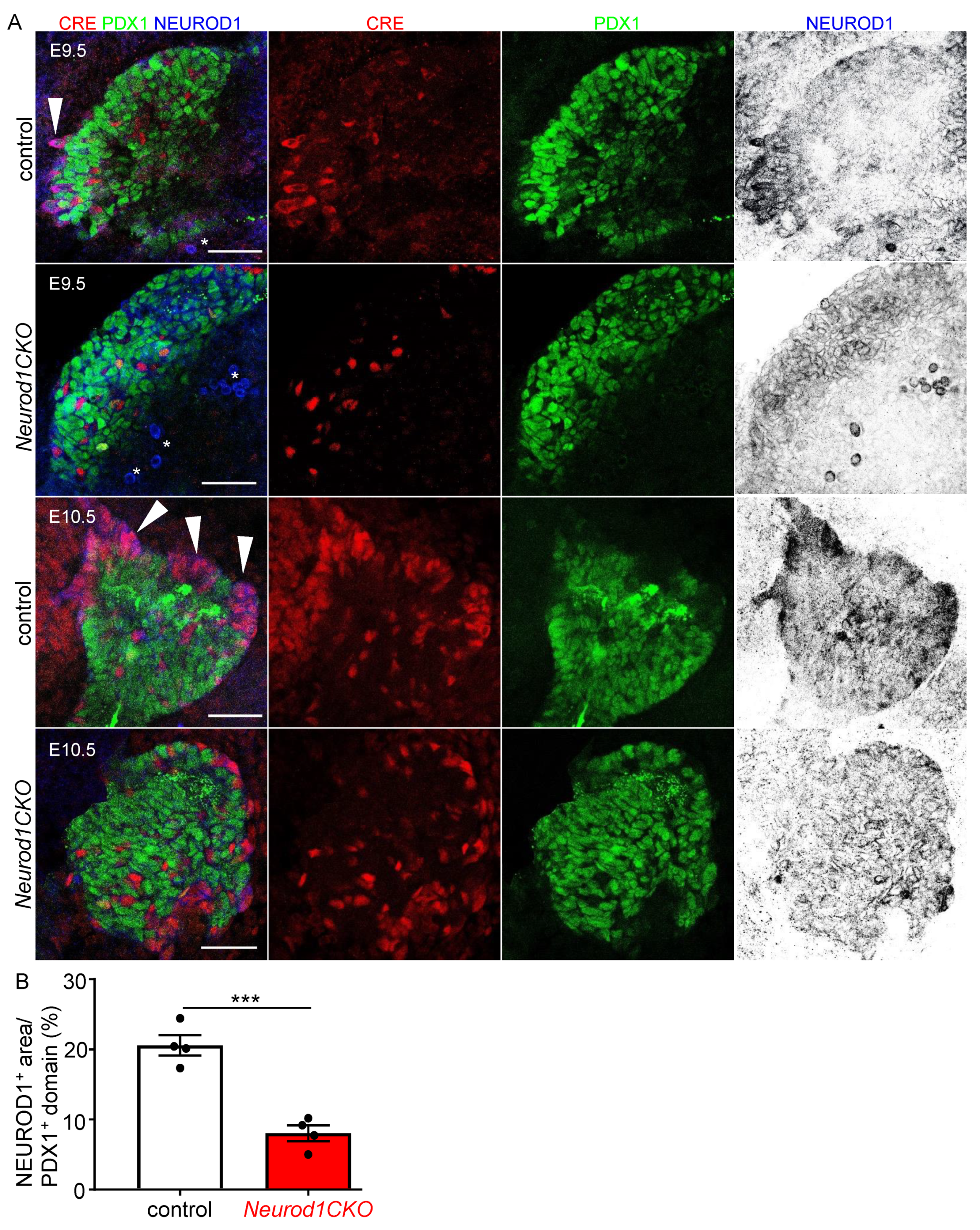
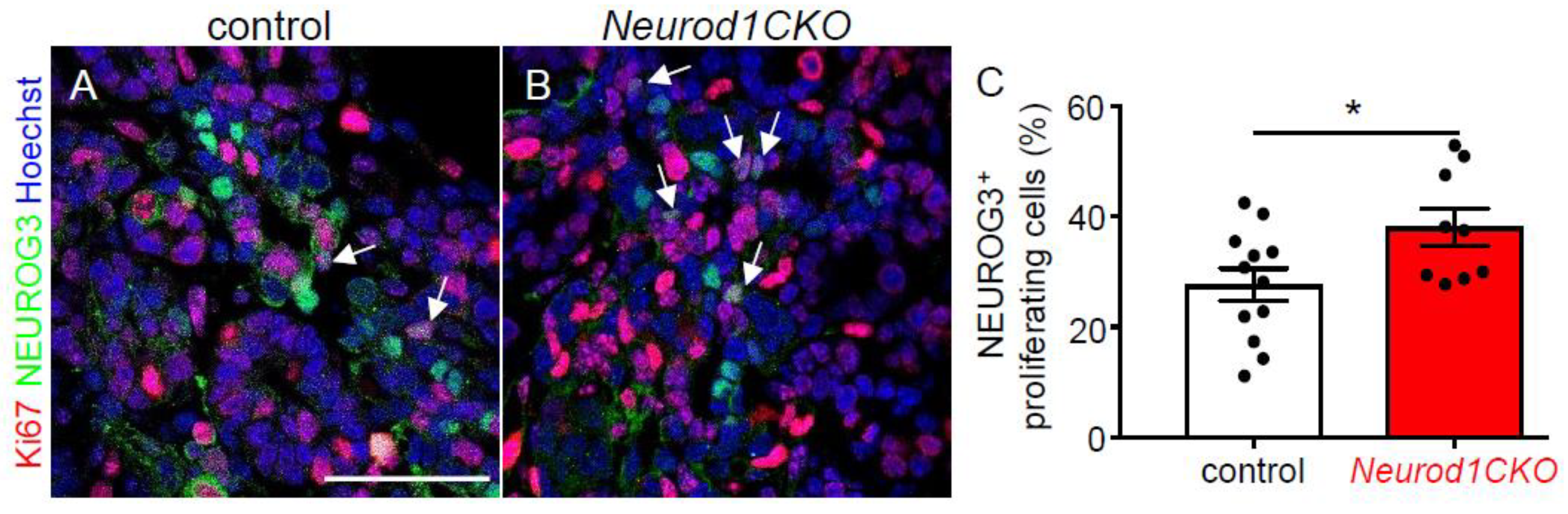
2.2. Altered Formation of Endocrine Cells in Neurod1CKO during the Primary Transition of Pancreas Development Corresponds to NEUROD1 Elimination
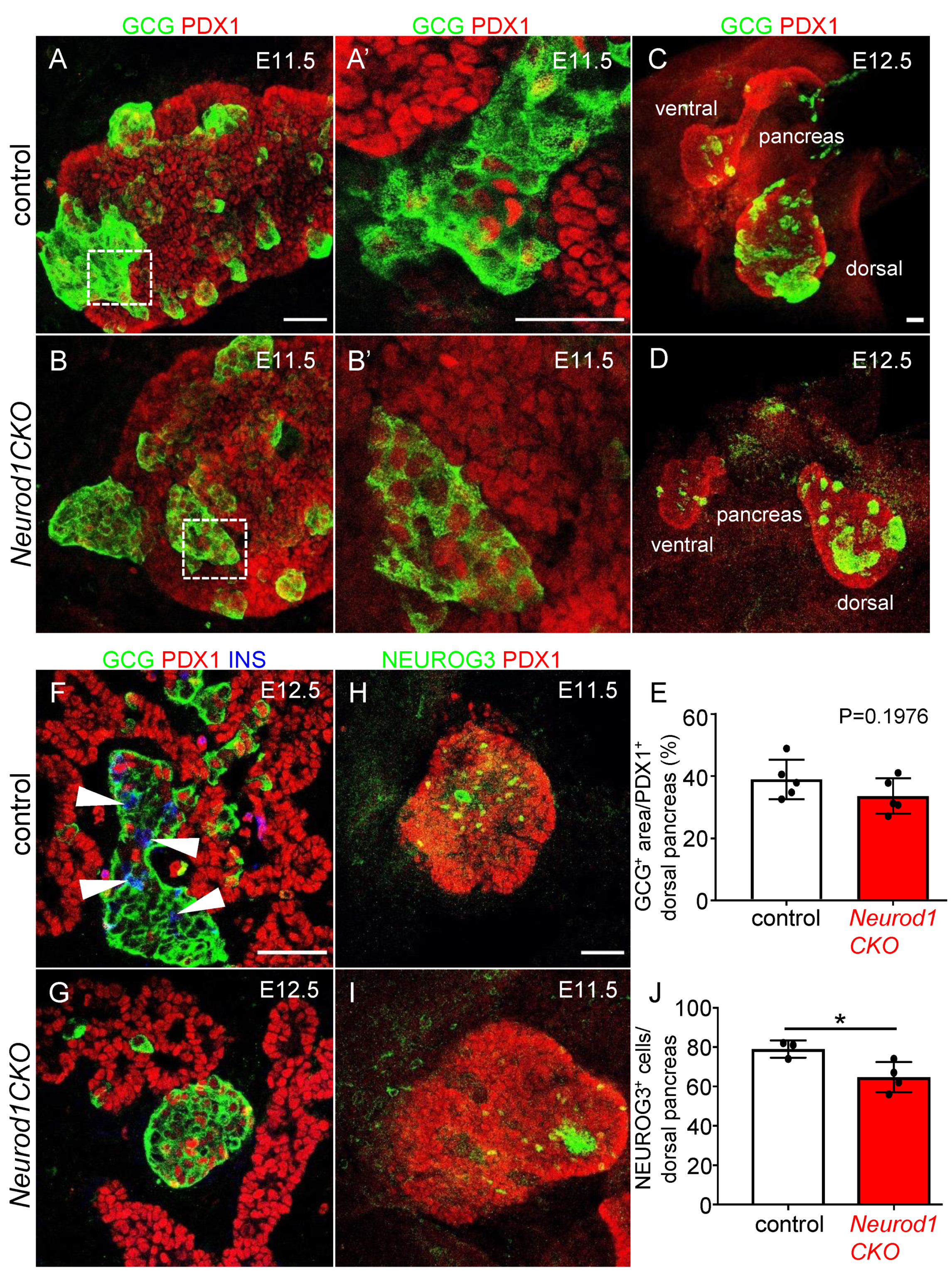
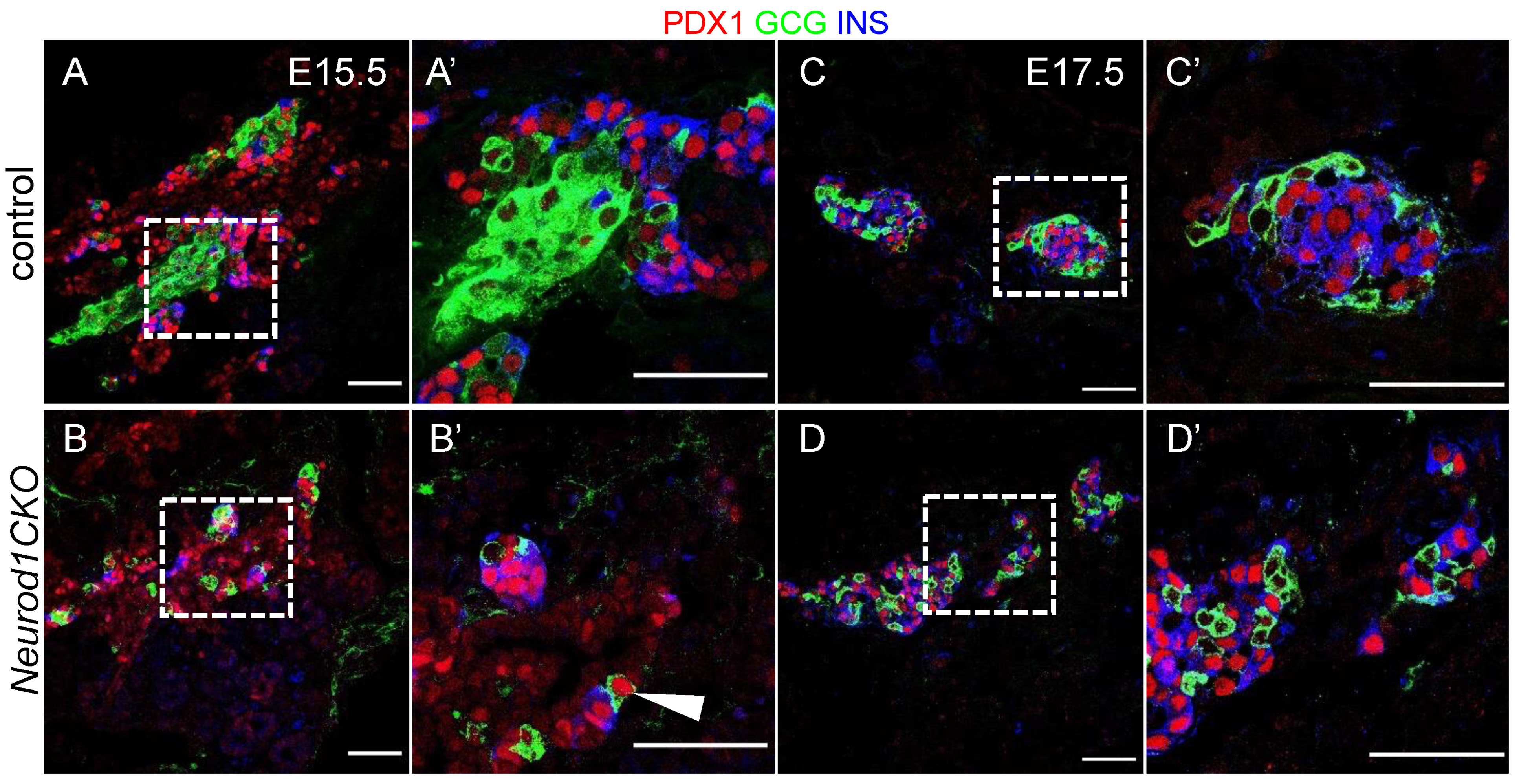
2.3. Differentiation of α and β Cell Lineages Is Affected in the Neurod1CKO Pancreas during the Early Phase of the Secondary Transition
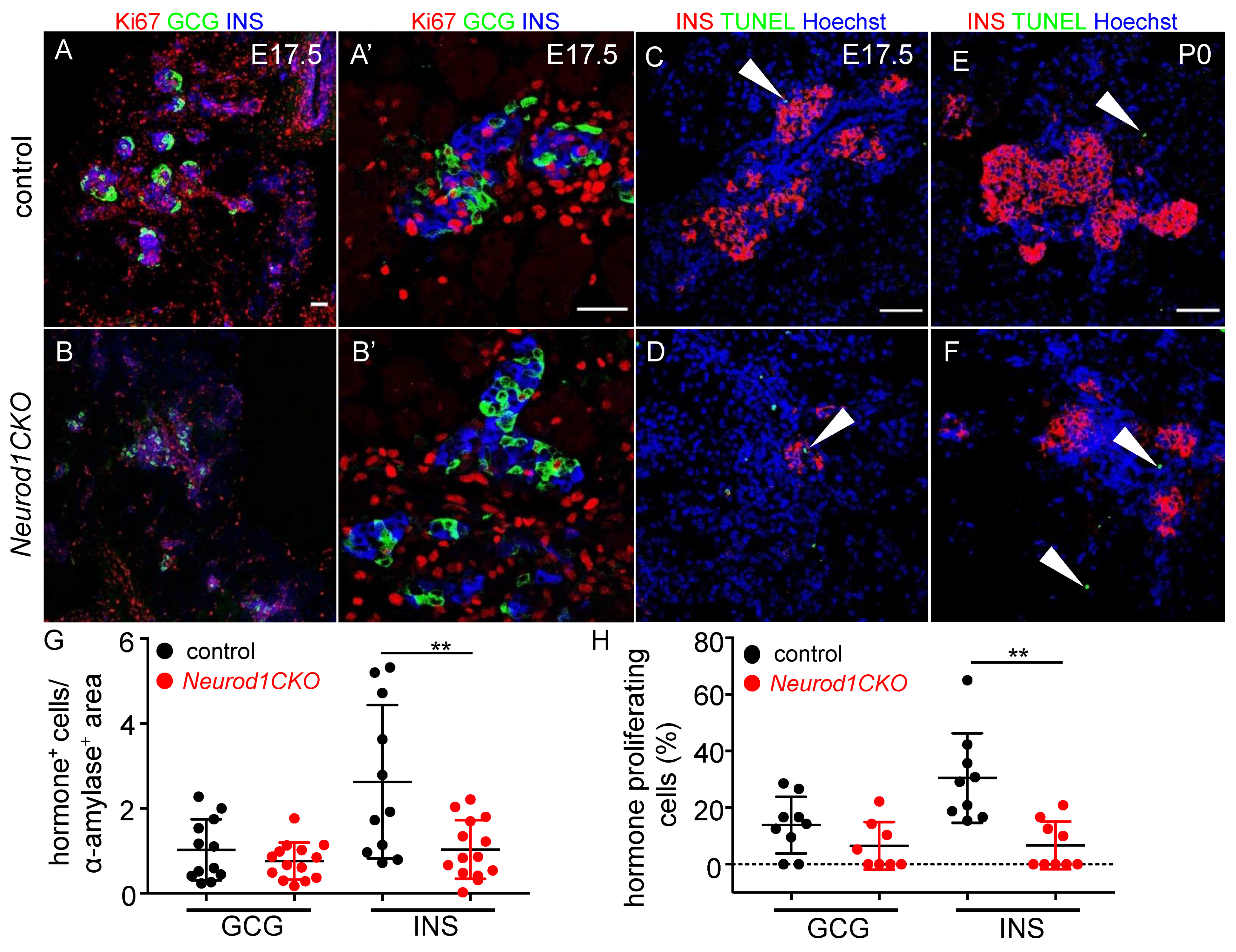
2.4. Deletion of Neurod1 Is Associated with Reduced Insulin Production, Disorganized Architecture of Islets of Langerhans, and Reduced Proliferation of β Cells
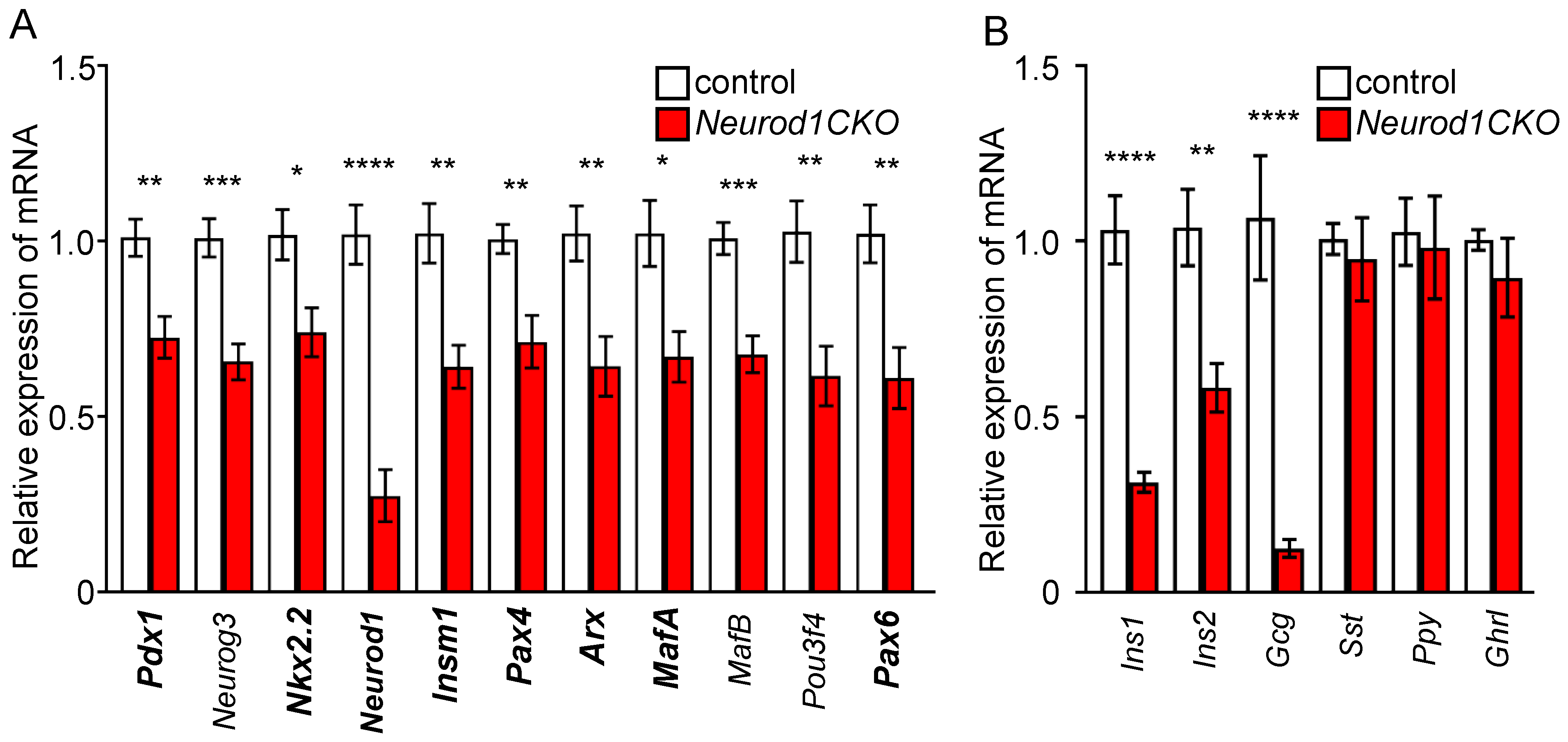
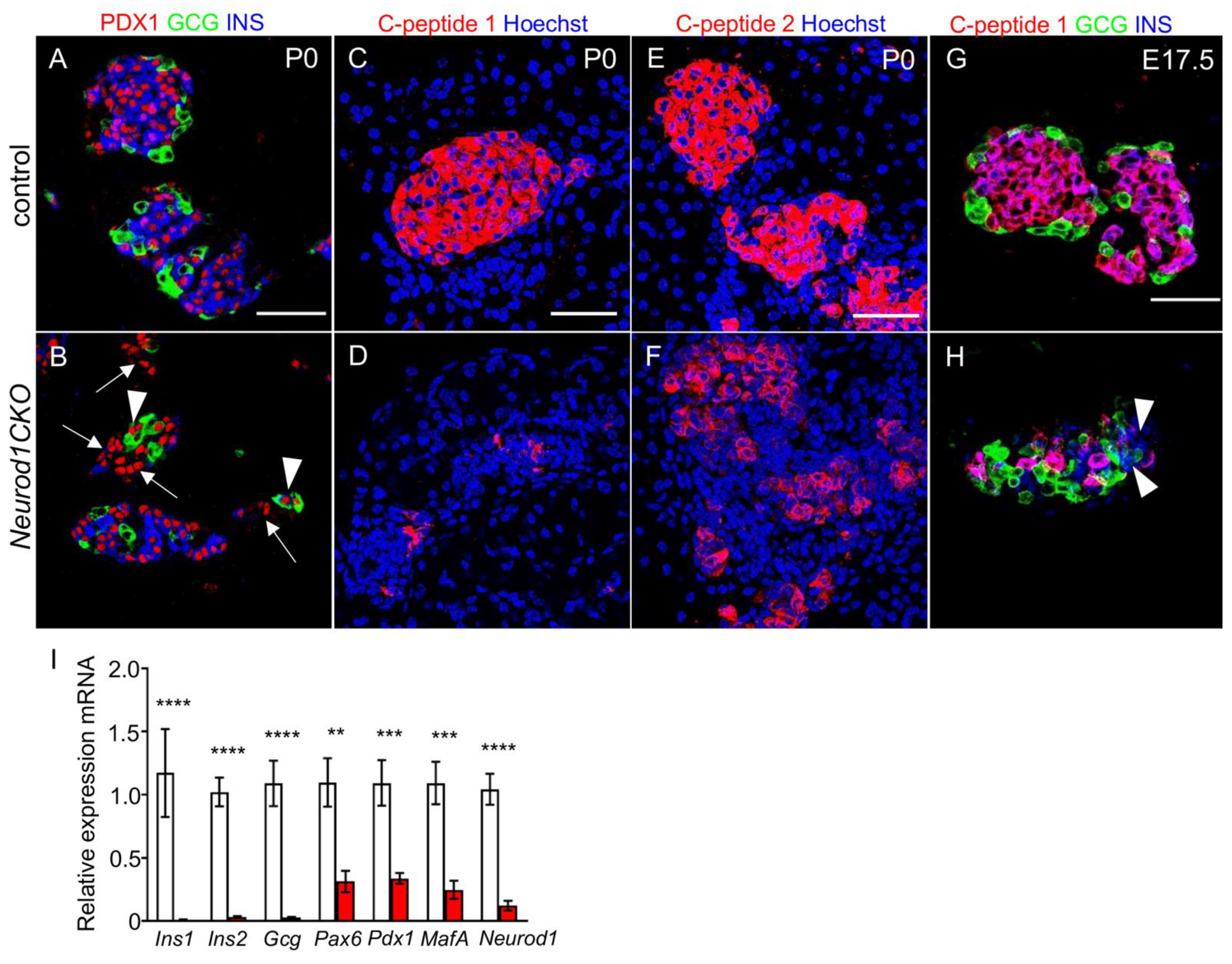
2.5. Abnormalities in the Expression of Pancreatic Endocrine Markers Are Associated with a Severe Diabetic Phenotype of Neurod1CKO after Birth
3. Discussion
4. Materials and Methods
4.1. Mouse Model
4.2. Immunohistochemistry
4.3. Reverse Transcription-Quantitative Real-Time Polymerase Chain Reaction
4.4. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, F.C.; Wright, C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011, 240, 530–565. [Google Scholar] [CrossRef]
- Bastidas-Ponce, A.; Scheibner, K.; Lickert, H.; Bakhti, M. Cellular and molecular mechanisms coordinating pancreas development. Development 2017, 144, 2873–2888. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [CrossRef]
- Herrera, P.L.; Huarte, J.; Sanvito, F.; Meda, P.; Orci, L.; Vassalli, J.D. Embryogenesis of the murine endocrine pancreas; early expression of pancreatic polypeptide gene. Development 1991, 113, 1257–1265. [Google Scholar] [CrossRef]
- Johansson, K.A.; Dursun, U.; Jordan, N.; Gu, G.; Beermann, F.; Gradwohl, G.; Grapin-Botton, A. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev. Cell 2007, 12, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Heller, R.S.; Jenny, M.; Collombat, P.; Mansouri, A.; Tomasetto, C.; Madsen, O.D.; Mellitzer, G.; Gradwohl, G.; Serup, P. Genetic determinants of pancreatic epsilon-cell development. Dev. Biol. 2005, 286, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Bastidas-Ponce, A.; Tritschler, S.; Dony, L.; Scheibner, K.; Tarquis-Medina, M.; Salinno, C.; Schirge, S.; Burtscher, I.; Bottcher, A.; Theis, F.J.; et al. Comprehensive single cell mRNA profiling reveals a detailed roadmap for pancreatic endocrinogenesis. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, N.; Chawla, R.; Mueller, J.; Vanderhooft, J.; Whitehorn, L.J.; Rosenthal, B.; Gurtler, M.; Estanboulieh, R.R.; Shvartsman, D.; Gifford, D.K.; et al. A Peninsular Structure Coordinates Asynchronous Differentiation with Morphogenesis to Generate Pancreatic Islets. Cell 2019, 176, 790–804.e13. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.L.; Zhang, Y.W.; Feng, Y.; Li, L.C.; Yang, L.; Xu, C.R. Deciphering Pancreatic Islet beta Cell and alpha Cell Maturation Pathways and Characteristic Features at the Single-Cell Level. Cell Metab. 2017, 25, 1194–1205.e4. [Google Scholar] [CrossRef] [Green Version]
- Jennings, R.E.; Scharfmann, R.; Staels, W. Transcription factors that shape the mammalian pancreas. Diabetologia 2020, 63, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Naya, F.J.; Huang, H.P.; Qiu, Y.; Mutoh, H.; DeMayo, F.J.; Leiter, A.B.; Tsai, M.J. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997, 11, 2323–2334. [Google Scholar] [CrossRef] [Green Version]
- Romer, A.I.; Singer, R.A.; Sui, L.; Egli, D.; Sussel, L. Murine Perinatal beta-Cell Proliferation and the Differentiation of Human Stem Cell-Derived Insulin-Expressing Cells Require NEUROD1. Diabetes 2019, 68, 2259–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malecki, M.T.; Jhala, U.S.; Antonellis, A.; Fields, L.; Doria, A.; Orban, T.; Saad, M.; Warram, J.H.; Montminy, M.; Krolewski, A.S. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat. Genet. 1999, 23, 323–328. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Minton, J.A.; Kantor, I.; Williams, D.; Ellard, S.; Hattersley, A.T. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 2010, 59, 2326–2331. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Motohashi, Y.; Yanagawa, T.; Maruyama, T.; Kasuga, A.; Hirose, H.; Matsubara, K.; Shimada, A.; Saruta, T. NeuroD/beta2 gene G-->A polymorphism may affect onset pattern of type 1 diabetes in Japanese. Diabetes Care 2001, 24, 1438–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Guo, M.; Huang, S.; Stein, R. Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, BETA2, and E47. Mol. Cell Biol. 2002, 22, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Stein, G.H.; Pan, N.; Goebbels, S.; Hornberg, H.; Nave, K.A.; Herrera, P.; White, P.; Kaestner, K.H.; Sussel, L.; et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010, 11, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Goebbels, S.; Bode, U.; Pieper, A.; Funfschilling, U.; Schwab, M.H.; Nave, K.A. Cre/loxP-mediated inactivation of the bHLH transcription factor gene NeuroD/BETA2. Genesis 2005, 42, 247–252. [Google Scholar] [CrossRef]
- Yang, L.; Cai, C.L.; Lin, L.; Qyang, Y.; Chung, C.; Monteiro, R.M.; Mummery, C.L.; Fishman, G.I.; Cogen, A.; Evans, S. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 2006, 133, 1575–1585. [Google Scholar] [CrossRef] [Green Version]
- Burlison, J.S.; Long, Q.; Fujitani, Y.; Wright, C.V.; Magnuson, M.A. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev. Biol. 2008, 316, 74–86. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.P.; Thorel, F.; Boyer, D.F.; Herrera, P.L.; Wright, C.V. Context-specific alpha- to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev. 2011, 25, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Artner, I.; Le Lay, J.; Hang, Y.; Elghazi, L.; Schisler, J.C.; Henderson, E.; Sosa-Pineda, B.; Stein, R. MafB: An activator of the glucagon gene expressed in developing islet alpha- and beta-cells. Diabetes 2006, 55, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosmain, Y.; Cheyssac, C.; Heddad Masson, M.; Dibner, C.; Philippe, J. Glucagon gene expression in the endocrine pancreas: The role of the transcription factor Pax6 in alpha-cell differentiation, glucagon biosynthesis and secretion. Diabetes Obes. Metab. 2011, 13 (Suppl. 1), 31–38. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Mansouri, A.; Hecksher-Sorensen, J.; Serup, P.; Krull, J.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, R.S.; Stoffers, D.A.; Liu, A.; Schedl, A.; Crenshaw, E.B., 3rd; Madsen, O.D.; Serup, P. The role of Brn4/Pou3f4 and Pax6 in forming the pancreatic glucagon cell identity. Dev. Biol. 2004, 268, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosmain, Y.; Katz, L.S.; Masson, M.H.; Cheyssac, C.; Poisson, C.; Philippe, J. Pax6 is crucial for beta-cell function, insulin biosynthesis, and glucose-induced insulin secretion. Mol. Endocrinol. 2012, 26, 696–709. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Ivanov, A.; Blasevic, D.; Muller, T.; Purfurst, B.; Sun, W.; Chen, W.; Poy, M.N.; Rajewsky, N.; Birchmeier, C. Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic beta-cell function. EMBO J. 2015, 34, 1417–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Mastracci, T.L.; Wilcox, C.L.; Arnes, L.; Panea, C.; Golden, J.A.; May, C.L.; Sussel, L. Nkx2.2 and Arx genetically interact to regulate pancreatic endocrine cell development and endocrine hormone expression. Dev. Biol. 2011, 359, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Osipovich, A.B.; Long, Q.; Manduchi, E.; Gangula, R.; Hipkens, S.B.; Schneider, J.; Okubo, T.; Stoeckert, C.J., Jr.; Takada, S.; Magnuson, M.A. Insm1 promotes endocrine cell differentiation by modulating the expression of a network of genes that includes Neurog3 and Ripply3. Development 2014, 141, 2939–2949. [Google Scholar] [CrossRef] [Green Version]
- Tennant, B.R.; Robertson, A.G.; Kramer, M.; Li, L.; Zhang, X.; Beach, M.; Thiessen, N.; Chiu, R.; Mungall, K.; Whiting, C.J.; et al. Identification and analysis of murine pancreatic islet enhancers. Diabetologia 2013, 56, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Banerjee, A.; Herring, C.A.; Attalla, J.; Hu, R.; Xu, Y.; Shao, Q.; Simmons, A.J.; Dadi, P.K.; Wang, S.; et al. Neurog3-Independent Methylation Is the Earliest Detectable Mark Distinguishing Pancreatic Progenitor Identity. Dev. Cell 2019, 48, 49–63.e7. [Google Scholar] [CrossRef]
- Duvillie, B.; Cordonnier, N.; Deltour, L.; Dandoy-Dron, F.; Itier, J.M.; Monthioux, E.; Jami, J.; Joshi, R.L.; Bucchini, D. Phenotypic alterations in insulin-deficient mutant mice. Proc. Natl. Acad. Sci. USA 1997, 94, 5137–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroux, L.; Desbois, P.; Lamotte, L.; Duvillie, B.; Cordonnier, N.; Jackerott, M.; Jami, J.; Bucchini, D.; Joshi, R.L. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 2001, 50 (Suppl. 1), S150. [Google Scholar] [CrossRef] [Green Version]
- Artner, I.; Hang, Y.; Mazur, M.; Yamamoto, T.; Guo, M.; Lindner, J.; Magnuson, M.A.; Stein, R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 2010, 59, 2530–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krentz, N.A.J.; Lee, M.Y.Y.; Xu, E.E.; Sproul, S.L.J.; Maslova, A.; Sasaki, S.; Lynn, F.C. Single-Cell Transcriptome Profiling of Mouse and hESC-Derived Pancreatic Progenitors. Stem Cell Rep. 2018, 11, 1551–1564. [Google Scholar] [CrossRef] [Green Version]
- Mastracci, T.L.; Anderson, K.R.; Papizan, J.B.; Sussel, L. Regulation of Neurod1 contributes to the lineage potential of Neurogenin3+ endocrine precursor cells in the pancreas. PLoS Genet. 2013, 9, e1003278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.S.; Loomis, Z.L.; Lee, J.E.; Sussel, L. Genetic identification of a novel NeuroD1 function in the early differentiation of islet alpha, PP and epsilon cells. Dev. Biol. 2007, 312, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Schonhoff, S.E.; Giel-Moloney, M.; Leiter, A.B. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev. Biol. 2004, 270, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000, 127, 2317–2322. [Google Scholar] [CrossRef]
- Gannon, M.; Ray, M.K.; Van Zee, K.; Rausa, F.; Costa, R.H.; Wright, C.V. Persistent expression of HNF6 in islet endocrine cells causes disrupted islet architecture and loss of beta cell function. Development 2000, 127, 2883–2895. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.J.; Sussel, L. Nkx2.2 regulates beta-cell function in the mature islet. Diabetes 2007, 56, 1999–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, A.; Hunter, C.S.; Murray, J.; Noble, D.; Cai, C.L.; Evans, S.M.; Stein, R.; May, C.L. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes 2009, 58, 2059–2069. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, U.; Pfaff, S.L.; Jessell, T.M.; Edlund, T.; Edlund, H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature 1997, 385, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Bohuslavova, R.; Cerychova, R.; Papousek, F.; Olejnickova, V.; Bartos, M.; Gorlach, A.; Kolar, F.; Sedmera, D.; Semenza, G.L.; Pavlinkova, G. HIF-1alpha is required for development of the sympathetic nervous system. Proc. Natl. Acad. Sci. USA 2019, 116, 13414–13423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerychova, R.; Bohuslavova, R.; Papousek, F.; Sedmera, D.; Abaffy, P.; Benes, V.; Kolar, F.; Pavlinkova, G. Adverse effects of Hif1a mutation and maternal diabetes on the offspring heart. Cardiovasc. Diabetol. 2018, 17, 68. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohuslavova, R.; Smolik, O.; Malfatti, J.; Berkova, Z.; Novakova, Z.; Saudek, F.; Pavlinkova, G. NEUROD1 Is Required for the Early α and β Endocrine Differentiation in the Pancreas. Int. J. Mol. Sci. 2021, 22, 6713. https://doi.org/10.3390/ijms22136713
Bohuslavova R, Smolik O, Malfatti J, Berkova Z, Novakova Z, Saudek F, Pavlinkova G. NEUROD1 Is Required for the Early α and β Endocrine Differentiation in the Pancreas. International Journal of Molecular Sciences. 2021; 22(13):6713. https://doi.org/10.3390/ijms22136713
Chicago/Turabian StyleBohuslavova, Romana, Ondrej Smolik, Jessica Malfatti, Zuzana Berkova, Zaneta Novakova, Frantisek Saudek, and Gabriela Pavlinkova. 2021. "NEUROD1 Is Required for the Early α and β Endocrine Differentiation in the Pancreas" International Journal of Molecular Sciences 22, no. 13: 6713. https://doi.org/10.3390/ijms22136713