
How Single-Molecule Localization Microscopy Expanded Our Mechanistic Understanding of RNA Polymerase II Transcription


Abstract
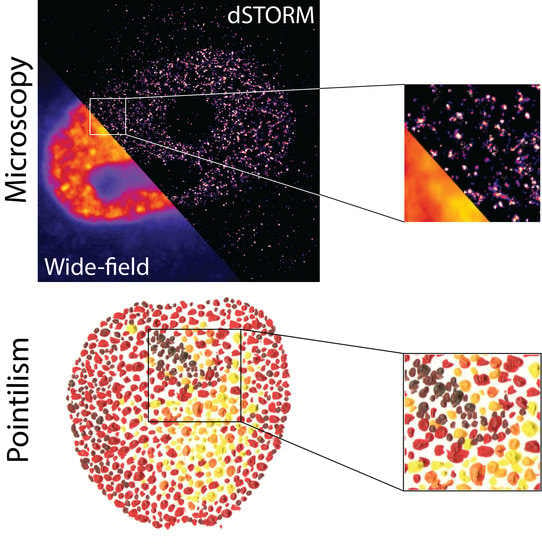
:
1. Introduction
2. Brief Introduction to SRM
2.1. Deterministic SRM Approaches: SIM and STED
2.2. Stochastic SRM Approaches: Single-Molecule Localization Microscopy
3. Progressive Development of Fluorophores for SMLM
4. Revised Model of Transcription Pre-Initiation Based on the TF Single-Molecule Kinetics
5. Dynamics of RNAPII by SMLM

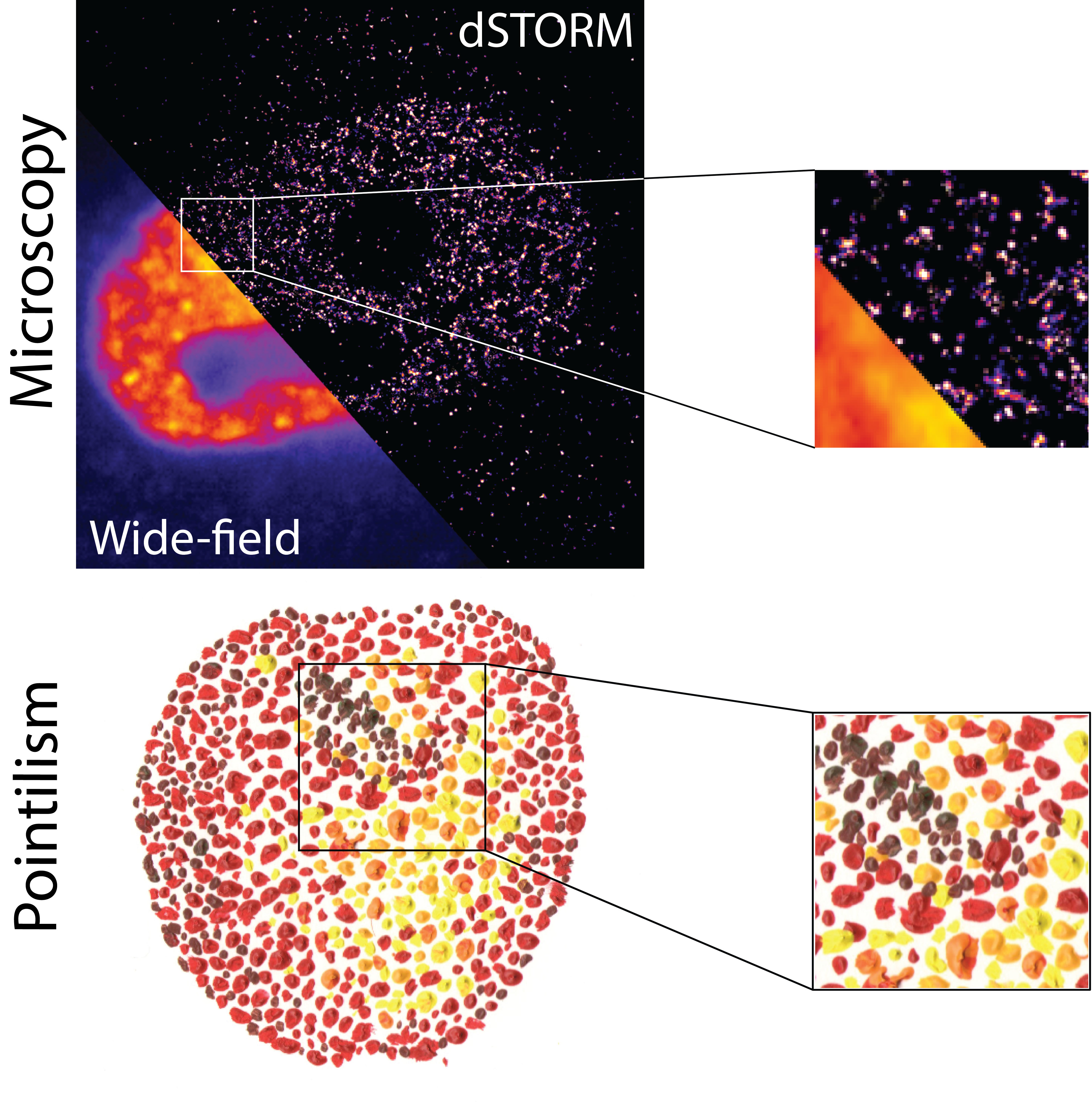{kind=link}
{kind=link}
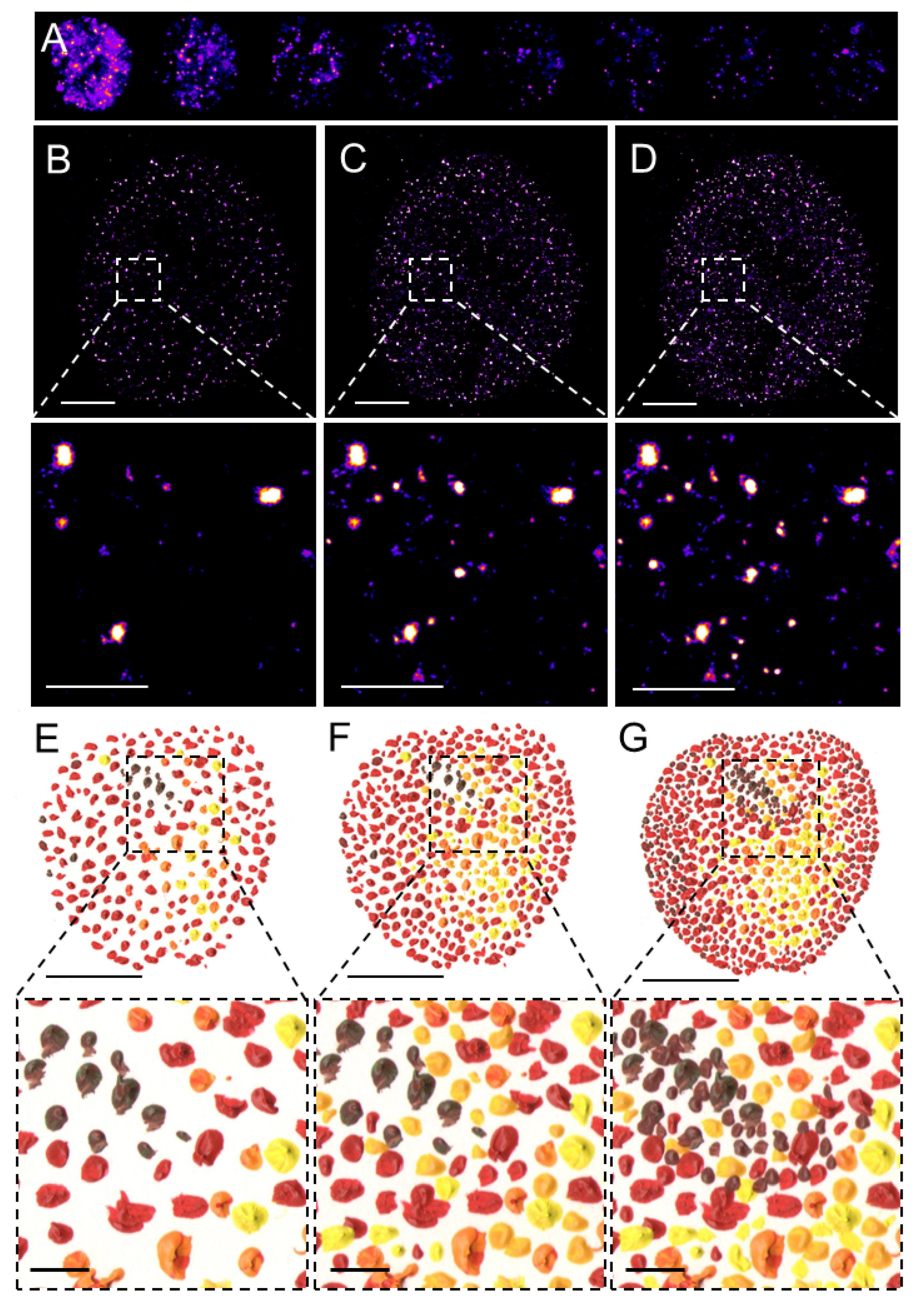{kind=link}
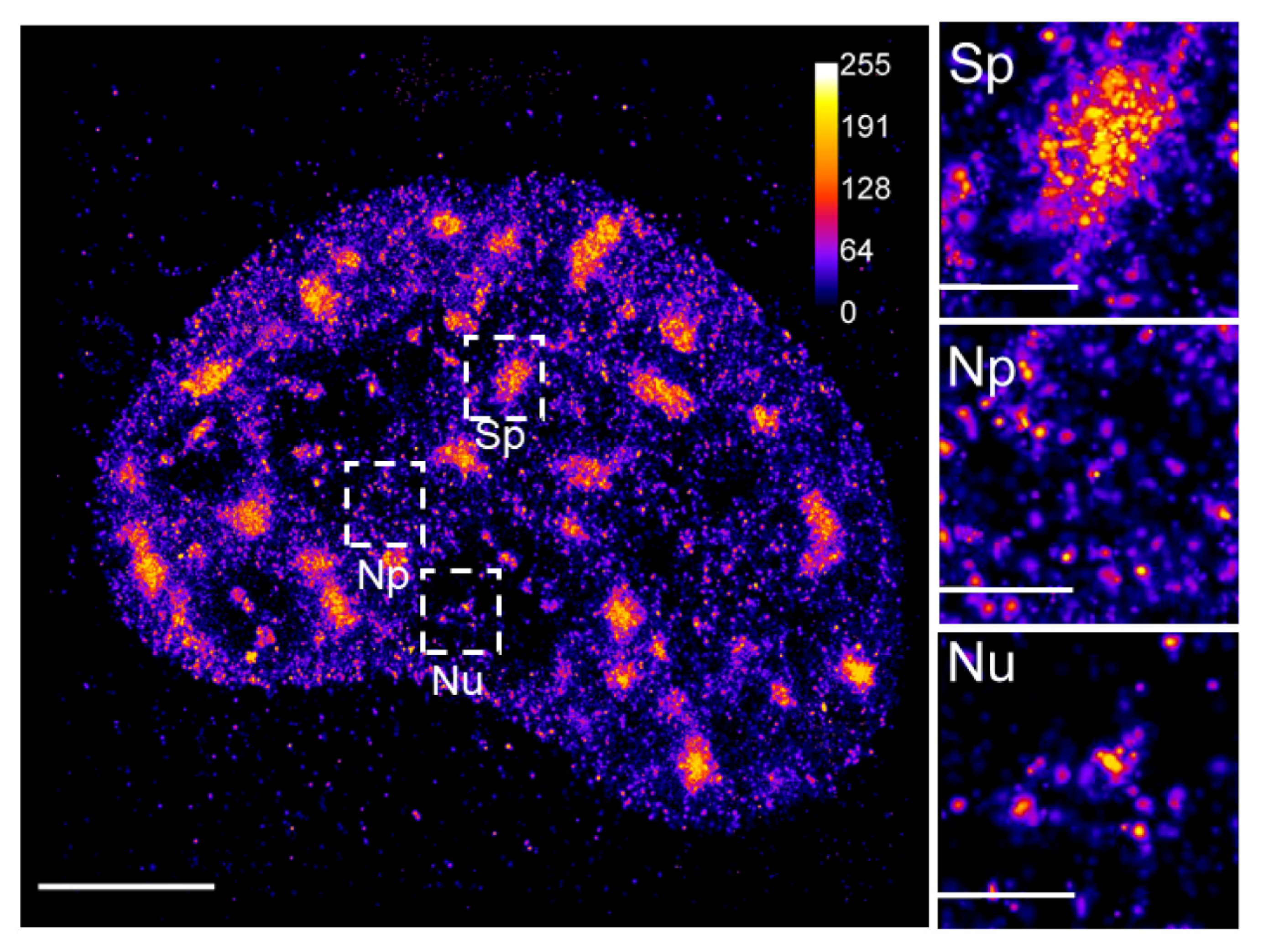{kind=link}
| Imaging Approach | Utilized Fluorophore | Target Molecule | Model System | Reference |
|---|---|---|---|---|
| HILO, SPT | 3xHaloTag-JF549 | Sox2 | mESCs | [78] |
| BBSP 2D SPT MFM 3D SPT | HaloTag-TMR | Sox2, Oct4 | mESCs 3T3 cell line | [72] |
| PALM, SPT | Dendra2 | c-Myc, P-TEFb | U-2 OS cell line | [67] |
| LLS, SPT, PCC | HaloTag-JF549 | Sox2 | mESCs | [92] |
| RLS, RT | mEos2 | GR | MCF-7 cell line | [47] |
| LLS tcPALM | Halo-JF549/646 | RPB1 Med | mESCs | [66] |
| PALM | Dendra2 HaloTag-JF549 | RPB1 Sox2 | mESCs | [92] |
| tcPALM | Dendra2 | RPB1 | U-2 OS cell line MEFs | [63] [64,65] |
| SPA-SPT | Halo-PA-JF549 | RPB1 | U-2 OS cell line | [103] |
| RLS | SNAP-TMR | RPB1 | U-2 OS cell line | [48] |
| BLM | Dendra2 | RPB1 | U-2 OS cell line | [108] |
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Archiv Für Mikrosk. Anatomie 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Cremer, C.; Szczurek, A.; Schock, F.; Gourram, A.; Birk, U. Super-resolution microscopy approaches to nuclear nanostructure imaging. Methods 2017, 123, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Rayleig, L. On the Theory of Optical Images, with special reference to the Microscope. J. R. Microsc. Soc. 1896, 23, 474–482. [Google Scholar] [CrossRef]
- Pombo, A.; Hollinshead, M.; Cook, P.R. Bridging the resolution gap: Imaging the same transcription factories in cryosections by light and electron microscopy. J. Histochem. Cytochem. 1999, 47, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedojadlo, J.; Perret-Vivancos, C.; Kalland, K.H.; Cmarko, D.; Cremer, T.; van Driel, R.; Fakan, S. Transcribed DNA is preferentially located in the perichromatin region of mammalian cell nuclei. Exp. Cell Res. 2011, 317, 433–444. [Google Scholar] [CrossRef]
- Rouquette, J.; Cremer, C.; Cremer, T.; Fakan, S. Functional nuclear architecture studied by microscopy: Present and future. Int. Rev. Cell Mol. Biol. 2010, 282, 1–90. [Google Scholar] [CrossRef]
- Hanske, J.; Sadian, Y.; Müller, C.W. The cryo-EM resolution revolution and transcription complexes. Curr. Opin. Struct. Biol. 2018, 52, 8–15. [Google Scholar] [CrossRef]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [Green Version]
- Hell, S.W. Microscopy and its focal switch. Nat. Methods 2009, 6, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hell, S.W. Toward fluorescence nanoscopy. Nat. Biotechnol. 2003, 21, 1347–1355. [Google Scholar] [CrossRef]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Merino, D. Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J. Neurochem. 2015, 135, 643–658. [Google Scholar] [CrossRef]
- Sahl, S.J.; Hell, S.W. High-Resolution 3D Light Microscopy with STED and RESOLFT. In High Resolution Imaging in Microscopy and Ophthalmology: New Frontiers in Biomedical Optics; Bille, J.F., Ed.; Heidelberg University: Heidelberg, Germany, 2019; pp. 3–32. [Google Scholar]
- Hofmann, M.; Eggeling, C.; Jakobs, S.; Hell, S.W. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 17565–17569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.G. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 2005, 102, 13081–13086. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Bailey, B.; Farkas, D.L.; Taylor, D.L.; Lanni, F. Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature 1993, 366, 44–48. [Google Scholar] [CrossRef]
- Gustafsson, M.G.; Shao, L.; Carlton, P.M.; Wang, C.J.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [Green Version]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Baddeley, D.; Chagin, V.O.; Schermelleh, L.; Martin, S.; Pombo, A.; Carlton, P.M.; Gahl, A.; Domaing, P.; Birk, U.; Leonhardt, H.; et al. Measurement of replication structures at the nanometer scale using super-resolution light microscopy. Nucleic Acids Res. 2010, 38, e8. [Google Scholar] [CrossRef] [Green Version]
- Kner, P.; Chhun, B.B.; Griffis, E.R.; Winoto, L.; Gustafsson, M.G. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 2009, 6, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [Green Version]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Heilemann, M.; van de Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008, 47, 6172–6176. [Google Scholar] [CrossRef]
- Wombacher, R.; Heidbreder, M.; van de Linde, S.; Sheetz, M.P.; Heilemann, M.; Cornish, V.W.; Sauer, M. Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods 2010, 7, 717–719. [Google Scholar] [CrossRef]
- van de Linde, S.; Löschberger, A.; Klein, T.; Heidbreder, M.; Wolter, S.; Heilemann, M.; Sauer, M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011, 6, 991–1009. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Patterson, G.H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009, 19, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Suárez, M.; Ting, A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 929–943. [Google Scholar] [CrossRef]
- Lidke, K.; Rieger, B.; Jovin, T.; Heintzmann, R. Superresolution by localization of quantum dots using blinking statistics. Opt. Express 2005, 13, 7052–7062. [Google Scholar] [CrossRef]
- Huang, B.; Jones, S.A.; Brandenburg, B.; Zhuang, X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 2008, 5, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Shim, S.H.; He, J.; Zhuang, X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods 2011, 8, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Legant, W.R.; Shao, L.; Grimm, J.B.; Brown, T.A.; Milkie, D.E.; Avants, B.B.; Lavis, L.D.; Betzig, E. High-density three-dimensional localization microscopy across large volumes. Nat. Methods 2016, 13, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Juette, M.F.; Gould, T.J.; Lessard, M.D.; Mlodzianoski, M.J.; Nagpure, B.S.; Bennett, B.T.; Hess, S.T.; Bewersdorf, J. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 2008, 5, 527–529. [Google Scholar] [CrossRef]
- Pavani, S.R.; Thompson, M.A.; Biteen, J.S.; Lord, S.J.; Liu, N.; Twieg, R.J.; Piestun, R.; Moerner, W.E. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc. Natl. Acad. Sci. USA 2009, 106, 2995–2999. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Lavis, L.D.; Betzig, E. Imaging live-cell dynamics and structure at the single-molecule level. Mol. Cell 2015, 58, 644–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Traffic 2001, 2, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, P.; de Medeiros, G.; Balázs, B.; Norlin, N.; Besir, C.; Hanne, J.; Kräusslich, H.G.; Engelhardt, J.; Sahl, S.J.; Hell, S.W.; et al. Breaking the diffraction limit of light-sheet fluorescence microscopy by RESOLFT. Proc. Natl. Acad. Sci. USA 2016, 113, 3442–3446. [Google Scholar] [CrossRef] [Green Version]
- Planchon, T.A.; Gao, L.; Milkie, D.E.; Davidson, M.W.; Galbraith, J.A.; Galbraith, C.G.; Betzig, E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods 2011, 8, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Shao, L.; Chen, B.C.; Betzig, E. 3D live fluorescence imaging of cellular dynamics using Bessel beam plane illumination microscopy. Nat. Protoc. 2014, 9, 1083–1101. [Google Scholar] [CrossRef]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., 3rd; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, J.C.; Suter, D.M.; Roy, R.; Zhao, Z.W.; Chapman, A.R.; Basu, S.; Maniatis, T.; Xie, X.S. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat. Methods 2013, 10, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.W.; Roy, R.; Gebhardt, J.C.; Suter, D.M.; Chapman, A.R.; Xie, X.S. Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Eilers, Y.; Ta, H.; Gwosch, K.C.; Balzarotti, F.; Hell, S.W. MINFLUX monitors rapid molecular jumps with superior spatiotemporal resolution. Proc. Natl. Acad. Sci. USA 2018, 115, 6117–6122. [Google Scholar] [CrossRef] [Green Version]
- Gwosch, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef]
- Schmidt, R.; Weihs, T.; Wurm, C.A.; Jansen, I.; Rehman, J.; Sahl, S.J.; Hell, S.W. MINFLUX nanometer-scale 3D imaging and microsecond-range tracking on a common fluorescence microscope. Nat. Commun. 2021, 12, 1478. [Google Scholar] [CrossRef]
- Dempsey, G.T.; Vaughan, J.C.; Chen, K.H.; Bates, M.; Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 2011, 8, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; Tkachuk, A.N.; Xie, L.; Choi, H.; Mohar, B.; Falco, N.; Schaefer, K.; Patel, R.; Zheng, Q.; Liu, Z.; et al. A general method to optimize and functionalize red-shifted rhodamine dyes. Nat. Methods 2020, 17, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Lardon, N.; Wang, L.; Tschanz, A.; Hoess, P.; Tran, M.; D’Este, E.; Ries, J.; Johnsson, K. Systematic Tuning of Rhodamine Spirocyclization for Super-Resolution Microscopy. bioRxiv 2021. [Google Scholar] [CrossRef]
- McKinney, S.A.; Murphy, C.S.; Hazelwood, K.L.; Davidson, M.W.; Looger, L.L. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 2009, 6, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Röcker, C.; Salih, A.; Spindler, K.D.; Nienhaus, G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurskaya, N.G.; Verkhusha, V.V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Lukyanov, S.; Lukyanov, K.A. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat. Protoc. 2007, 2, 2024–2032. [Google Scholar] [CrossRef]
- Zhang, L.; Gurskaya, N.G.; Merzlyak, E.M.; Staroverov, D.B.; Mudrik, N.N.; Samarkina, O.N.; Vinokurov, L.M.; Lukyanov, S.; Lukyanov, K.A. Method for real-time monitoring of protein degradation at the single cell level. Biotechniques 2007, 42, 446–450. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Shin, J.Y.; Lee, A.; Bustamante, C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM). Proc. Natl. Acad. Sci. USA 2012, 109, 17436–17441. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, D.; Okumus, B.; Chien, P.; Baker, T.A.; Paulsson, J. Segregation of molecules at cell division reveals native protein localization. Nat. Methods 2012, 9, 480–482, Published 2012 Apr 8. [Google Scholar] [CrossRef] [Green Version]
- Cisse, I.I.; Izeddin, I.; Causse, S.Z.; Boudarene, L.; Senecal, A.; Muresan, L.; Dugast-Darzacq, C.; Hajj, B.; Dahan, M.; Darzacq, X. Real-time dynamics of RNA polymerase II clustering in live human cells. Science 2013, 341, 664–667. [Google Scholar] [CrossRef]
- Cho, W.K.; Jayanth, N.; English, B.P.; Inoue, T.; Andrews, J.O.; Conway, W.; Grimm, J.B.; Spille, J.H.; Lavis, L.D.; Lionnet, T.; et al. RNA Polymerase II cluster dynamics predict mRNA output in living cells. Elife 2016, 5, e13617. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.K.; Jayanth, N.; Mullen, S.; Tan, T.H.; Jung, Y.J.; Cissé, I.I. Super-resolution imaging of fluorescently labeled, endogenous RNA Polymerase II in living cells with CRISPR/Cas9-mediated gene editing. Sci. Rep. 2016, 6, 35949. [Google Scholar] [CrossRef]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Izeddin, I.; Récamier, V.; Bosanac, L.; Cissé, I.I.; Boudarene, L.; Dugast-Darzacq, C.; Proux, F.; Bénichou, O.; Voituriez, R.; Bensaude, O.; et al. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife 2014, 3, e02230. [Google Scholar] [CrossRef] [Green Version]
- Rollins, G.C.; Shin, J.Y.; Bustamante, C.; Pressé, S. Stochastic approach to the molecular counting problem in superresolution microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E110–E118. [Google Scholar] [CrossRef] [Green Version]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa IRJr Kindermann, M.; Beaufils, F.; Johnsson, K. An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Li, L.; Chen, B.C.; Revyakin, A.; Hajj, B.; Legant, W.; Dahan, M.; Lionnet, T.; Betzig, E.; et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 2014, 156, 1274–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presman, D.M.; Ball, D.A.; Paakinaho, V.; Grimm, J.B.; Lavis, L.D.; Karpova, T.S.; Hager, G.L. Quantifying transcription factor binding dynamics at the single-molecule level in live cells. Methods 2017, 123, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, J.B.; English, B.P.; Chen, J.; Slaughter, J.P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J.J.; Normanno, D.; Singer, R.H.; et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 2015, 12, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ayala, A.X.; Chung, I.; Weigel, A.V.; Ranjan, A.; Falco, N.; Grimm, J.B.; Tkachuk, A.N.; Wu, C.; Lippincott-Schwartz, J.; et al. Rational Design of Fluorogenic and Spontaneously Blinking Labels for Super-Resolution Imaging. ACS Cent. Sci. 2019, 5, 1602–1613, Erratum in ACS Cent. Sci. 2020, 6, 1844. [Google Scholar] [CrossRef] [PubMed]
- Grimm, J.B.; Muthusamy, A.K.; Liang, Y.; Brown, T.A.; Lemon, W.C.; Patel, R.; Lu, R.; Macklin, J.J.; Keller, P.J.; Ji, N.; et al. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Grimm, J.B.; English, B.P.; Choi, H.; Muthusamy, A.K.; Mehl, B.P.; Dong, P.; Brown, T.A.; Lippincott-Schwartz, J.; Liu, Z.; Lionnet, T.; et al. Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 2016, 13, 985–988. [Google Scholar] [CrossRef]
- Liu, H.; Dong, P.; Ioannou, M.S.; Li, L.; Shea, J.; Pasolli, H.A.; Grimm, J.B.; Rivlin, P.K.; Lavis, L.D.; Koyama, M.; et al. Visualizing long-term single-molecule dynamics in vivo by stochastic protein labeling. Proc. Natl. Acad. Sci. USA 2018, 115, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Binns, T.C.; Ayala, A.X.; Grimm, J.B.; Tkachuk, A.N.; Castillon, G.A.; Phan, S.; Zhang, L.; Brown, T.A.; Liu, Z.; Adams, S.R.; et al. Rational Design of Bioavailable Photosensitizers for Manipulation and Imaging of Biological Systems. Cell Chem. Biol. 2020, 27, 1063–1072.e7. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Gorski, S.A.; Dundr, M.; Misteli, T. The road much traveled: Trafficking in the cell nucleus. Curr. Opin. Cell Biol. 2006, 18, 284–290. [Google Scholar] [CrossRef]
- Hager, G.L.; McNally, J.G.; Misteli, T. Transcription dynamics. Mol. Cell 2009, 35, 741–753. [Google Scholar] [CrossRef]
- Woringer, M.; Darzacq, X. Protein motion in the nucleus: From anomalous diffusion to weak interactions. Biochem. Soc. Trans. 2018, 46, 945–956. [Google Scholar] [CrossRef] [Green Version]
- McNally, J.G.; Müller, W.G.; Walker, D.; Wolford, R.; Hager, G.L. The glucocorticoid receptor: Rapid exchange with regulatory sites in living cells. Science 2000, 287, 1262–1265. [Google Scholar] [CrossRef]
- Misteli, T. Protein dynamics: Implications for nuclear architecture and gene expression. Science 2001, 291, 843–847. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2001, 2, 898–907. [Google Scholar] [CrossRef]
- Darzacq, X.; Yao, J.; Larson, D.R.; Causse, S.Z.; Bosanac, L.; de Turris, V.; Ruda, V.M.; Lionnet, T.; Zenklusen, D.; Guglielmi, B.; et al. Imaging transcription in living cells. Annu. Rev. Biophys. 2009, 38, 173–196. [Google Scholar] [CrossRef] [Green Version]
- Fuda, N.J.; Ardehali, M.B.; Lis, J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 2009, 461, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J. Single-particle tracking photoactivated localization microscopy for mapping single-molecule dynamics. Methods Enzymol. 2010, 475, 109–120. [Google Scholar] [CrossRef]
- Hansen, A.S.; Woringer, M.; Grimm, J.B.; Lavis, L.D.; Tjian, R.; Darzacq, X. Robust model-based analysis of single-particle tracking experiments with Spot-On. Elife 2018, 7, e33125. [Google Scholar] [CrossRef]
- Abrahamsson, S.; Chen, J.; Hajj, B.; Stallinga, S.; Katsov, A.Y.; Wisniewski, J.; Mizuguchi, G.; Soule, P.; Mueller, F.; Dugast Darzacq, C.; et al. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat. Methods 2013, 10, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Legant, W.R.; Chen, B.C.; Li, L.; Grimm, J.B.; Lavis, L.D.; Betzig, E.; Tjian, R. 3D imaging of Sox2 enhancer clusters in embryonic stem cells. Elife 2014, 3, e04236. [Google Scholar] [CrossRef] [PubMed]
- Veatch, S.L.; Machta, B.B.; Shelby, S.A.; Chiang, E.N.; Holowka, D.A.; Baird, B.A. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS ONE 2012, 7, e31457. [Google Scholar] [CrossRef] [PubMed]
- Elf, J.; Li, G.W.; Xie, X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science 2007, 316, 1191–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprague, B.L.; Müller, F.; Pego, R.L.; Bungay, P.M.; Stavreva, D.A.; McNally, J.G. Analysis of binding at a single spatially localized cluster of binding sites by fluorescence recovery after photobleaching. Biophys. J. 2006, 91, 1169–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, J.B.; English, B.P.; Chen, J.; Slaughter, J.P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J.J.; Normanno, D.; Singer, R.H.; et al. Rapid dynamics of general transcription factor TFIIB binding during preinitiation complex assembly revealed by single-molecule analysis. Genes Dev. 2016, 30, 2106–2118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Revyakin, A.; Grimm, J.B.; Lavis, L.D.; Tjian, R. Single-molecule tracking of the transcription cycle by sub-second RNA detection. Elife 2014, 3, e01775. [Google Scholar] [CrossRef]
- Revyakin, A.; Zhang, Z.; Coleman, R.A.; Li, Y.; Inouye, C.; Lucas, J.K.; Park, S.R.; Chu, S.; Tjian, R. Transcription initiation by human RNA polymerase II visualized at single-molecule resolution. Genes Dev. 2012, 26, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- English, B.P.; Singer, R.H. A three-camera imaging microscope for high-speed single-molecule tracking and super-resolution imaging in living cells. Proc. SPIE Int. Soc. Opt. Eng. 2015, 9550, 955008. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.A.; Hassan, A.B.; Errington, R.J.; Cook, P.R. Visualization of focal sites of transcription within human nuclei. EMBO J. 1993, 12, 1059–1065. [Google Scholar] [CrossRef]
- Papantonis, A.; Cook, P.R. Transcription factories: Genome organization and gene regulation. Chem. Rev. 2013, 113, 8683–8705. [Google Scholar] [CrossRef]
- Cook, P.R. A model for all genomes: The role of transcription factories. J. Mol. Biol. 2010, 395, 1–10. [Google Scholar] [CrossRef]
- Hsin, J.P.; Manley, J.L. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012, 26, 2119–2137. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, M.; Hintermair, C.; Voß, K.; Eick, D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta 2013, 1829, 55–62. [Google Scholar] [CrossRef]
- LeBlanc, B.M.; Moreno, R.Y.; Escobar, E.E.; Venkat Ramani, M.K.; Brodbelt, J.S.; Zhang, Y. What’s all the phos about? Insights into the phosphorylation state of the RNA polymerase II C-terminal domain via mass spectrometry. RSC Chem. Biol. 2021. [Google Scholar] [CrossRef]
- Martin, R.D.; Hébert, T.E.; Tanny, J.C. Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. Int. J. Mol. Sci. 2020, 21, 3354. [Google Scholar] [CrossRef]
- Boehning, M.; Dugast-Darzacq, C.; Rankovic, M.; Hansen, A.S.; Yu, T.; Marie-Nelly, H.; McSwiggen, D.T.; Kokic, G.; Dailey, G.M.; Cramer, P.; et al. RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol. 2018, 25, 833–840. [Google Scholar] [CrossRef]
- Bertrand, E.; Chartrand, P.; Schaefer, M.; Shenoy, S.M.; Singer, R.H.; Long, R.M. Localization of ASH1 mRNA particles in living yeast. Mol. Cell 1998, 2, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Spille, J.H.; Hecht, M.; Grube, V.; Cho, W.K.; Lee, C.; Cissé, I.I. A CRISPR/Cas9 platform for MS2-labelling of single mRNA in live stem cells. Methods 2019, 153, 35–45. [Google Scholar] [CrossRef]
- Sawicka, A.; Villamil, G.; Lidschreiber, M.; Darzacq, X.; Dugast-Darzacq, C.; Schwalb, B.; Cramer, P. Transcription activation depends on the length of the RNA polymerase II C-terminal domain. EMBO J. 2021, 40, e107015. [Google Scholar] [CrossRef]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, 2555. [Google Scholar] [CrossRef] [Green Version]
- Cox, S.; Rosten, E.; Monypenny, J.; Jovanovic-Talisman, T.; Burnette, D.T.; Lippincott-Schwartz, J.; Jones, G.E.; Heintzmann, R. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat. Methods 2011, 9, 195–200. [Google Scholar] [CrossRef]
- Chen, X.; Wei, M.; Zheng, M.M.; Zhao, J.; Hao, H.; Chang, L.; Xi, P.; Sun, Y. Study of RNA Polymerase II Clustering inside Live-Cell Nuclei Using Bayesian Nanoscopy. ACS Nano 2016, 10, 2447–2454. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Bunce, M.W.; Bergendahl, K.; Anderson, R.A. Nuclear PI(4,5)P(2): A new place for an old signal. Biochim. Biophys. Acta 2006, 1761, 560–569. [Google Scholar] [CrossRef]
- Lewis, A.E.; Sommer, L.; Arntzen, M.Ø.; Strahm, Y.; Morrice, N.A.; Divecha, N.; D’Santos, C.S. Identification of nuclear phosphatidylinositol 4,5-bisphosphate-interacting proteins by neomycin extraction. Mol. Cell Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Sobol, M.; Krausová, A.; Yildirim, S.; Kalasová, I.; Fáberová, V.; Vrkoslav, V.; Philimonenko, V.; Marášek, P.; Pastorek, L.; Čapek, M.; et al. Nuclear phosphatidylinositol 4,5-bisphosphate islets contribute to efficient RNA polymerase II-dependent transcription. J. Cell Sci. 2018, 131, jcs211094. [Google Scholar] [CrossRef] [Green Version]
- Fáberová, V.; Kalasová, I.; Krausová, A.; Hozák, P. Super-Resolution Localisation of Nuclear PI(4)P and Identification of Its Interacting Proteome. Cells 2020, 9, 1191. [Google Scholar] [CrossRef]
- Shah, Z.H.; Jones, D.R.; Sommer, L.; Foulger, R.; Bultsma, Y.; D’Santos, C.; Divecha, N. Nuclear phosphoinositides and their impact on nuclear functions. FEBS J. 2013, 280, 6295–6310. [Google Scholar] [CrossRef] [PubMed]
- Cocco, L.; Follo, M.Y.; Manzoli, L.; Suh, P.G. Phosphoinositide-specific phospholipase C in health and disease. J. Lipid Res. 2015, 56, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sztacho, M.; Sobol, M.; Balaban, C.; Escudeiro Lopes, S.E.; Hozák, P. Nuclear phosphoinositides and phase separation: Important players in nuclear compartmentalization. Adv. Biol. Regul. 2019, 71, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Castano, E.; Yildirim, S.; Fáberová, V.; Krausová, A.; Uličná, L.; Paprčková, D.; Sztacho, M.; Hozák, P. Nuclear Phosphoinositides-Versatile Regulators of Genome Functions. Cells 2019, 8, 649. [Google Scholar] [CrossRef] [Green Version]
- Sztacho, M.; Šalovská, B.; Červenka, J.; Balaban, C.; Hoboth, P.; Hozák, P. Limited Proteolysis-Coupled Mass Spectrometry Identifies Phosphatidylinositol 4,5-Bisphosphate Effectors in Human Nuclear Proteome. Cells 2021, 10, 68. [Google Scholar] [CrossRef]
- Hoboth, P.; Sztacho, M.; Šebesta, O.; Schätz, M.; Castano, E.; Hozák, P. Nanoscale mapping of nuclear phosphatidylinositol phosphate landscape by dual-color dSTORM. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158890. [Google Scholar] [CrossRef]
- Hoboth, P.; Šebesta, O.; Sztacho, M.; Castano, E.; Hozak, P. Dual-color dSTORM imaging and ThunderSTORM image reconstruction and analysis to study the spatial organization of the nuclear phosphatidylinositol phosphates. MethodsX 2021, 101372. [Google Scholar] [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Franke, C.; Repnik, U.; Segeletz, S.; Brouilly, N.; Kalaidzidis, Y.; Verbavatz, J.M.; Zerial, M. Correlative single-molecule localization microscopy and electron tomography reveals endosome nanoscale domains. Traffic 2019, 20, 601–617. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoboth, P.; Šebesta, O.; Hozák, P. How Single-Molecule Localization Microscopy Expanded Our Mechanistic Understanding of RNA Polymerase II Transcription. Int. J. Mol. Sci. 2021, 22, 6694. https://doi.org/10.3390/ijms22136694
Hoboth P, Šebesta O, Hozák P. How Single-Molecule Localization Microscopy Expanded Our Mechanistic Understanding of RNA Polymerase II Transcription. International Journal of Molecular Sciences. 2021; 22(13):6694. https://doi.org/10.3390/ijms22136694
Chicago/Turabian StyleHoboth, Peter, Ondřej Šebesta, and Pavel Hozák. 2021. "How Single-Molecule Localization Microscopy Expanded Our Mechanistic Understanding of RNA Polymerase II Transcription" International Journal of Molecular Sciences 22, no. 13: 6694. https://doi.org/10.3390/ijms22136694