
Cross-Linking Effects Dictate the Preference of Galectins to Bind LacNAc-Decorated HPMA Copolymers

 ,
,  ,
,  , , ,
, , ,  , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
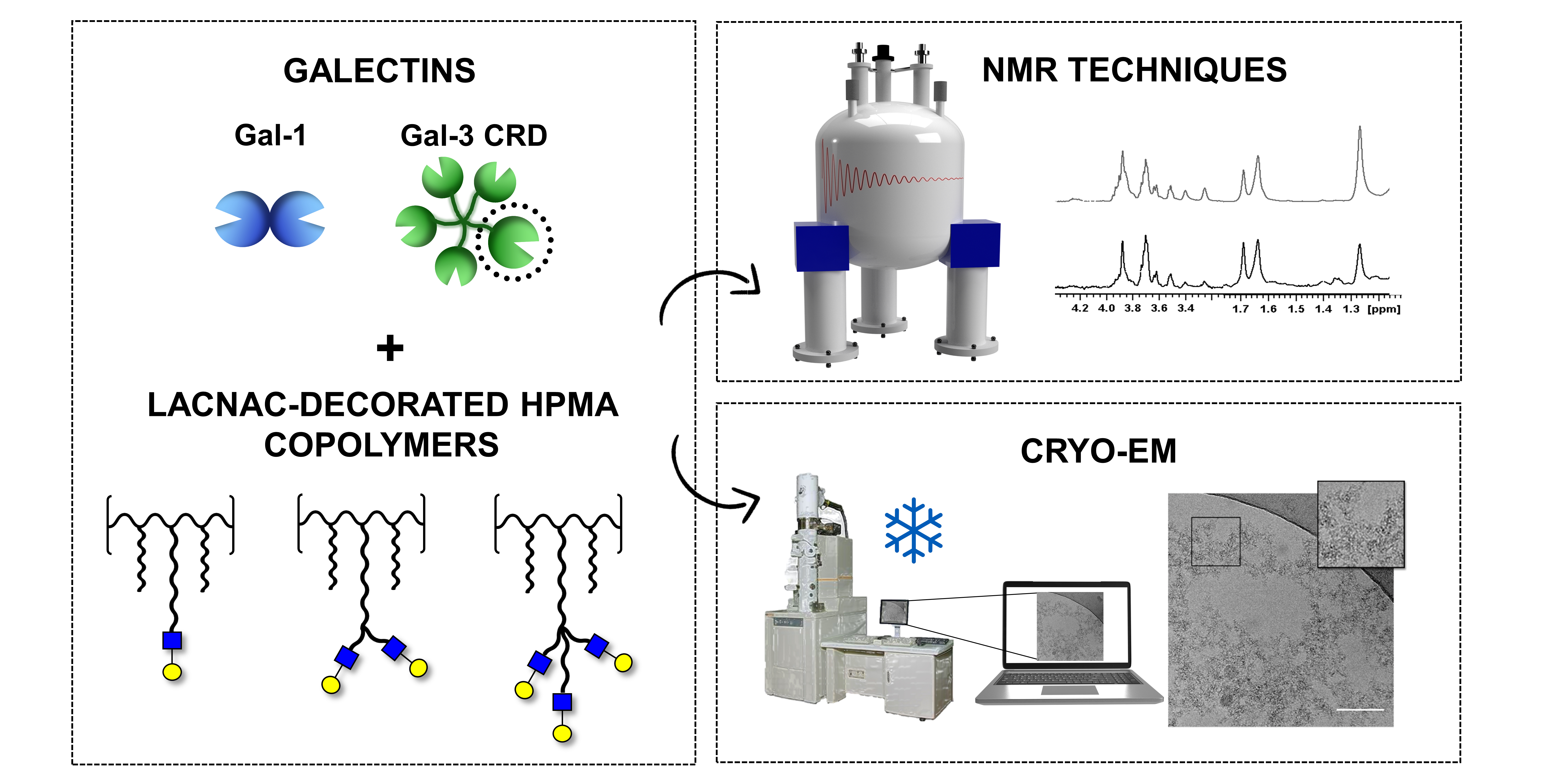
2.1. Synthesis of Ligands 2–6
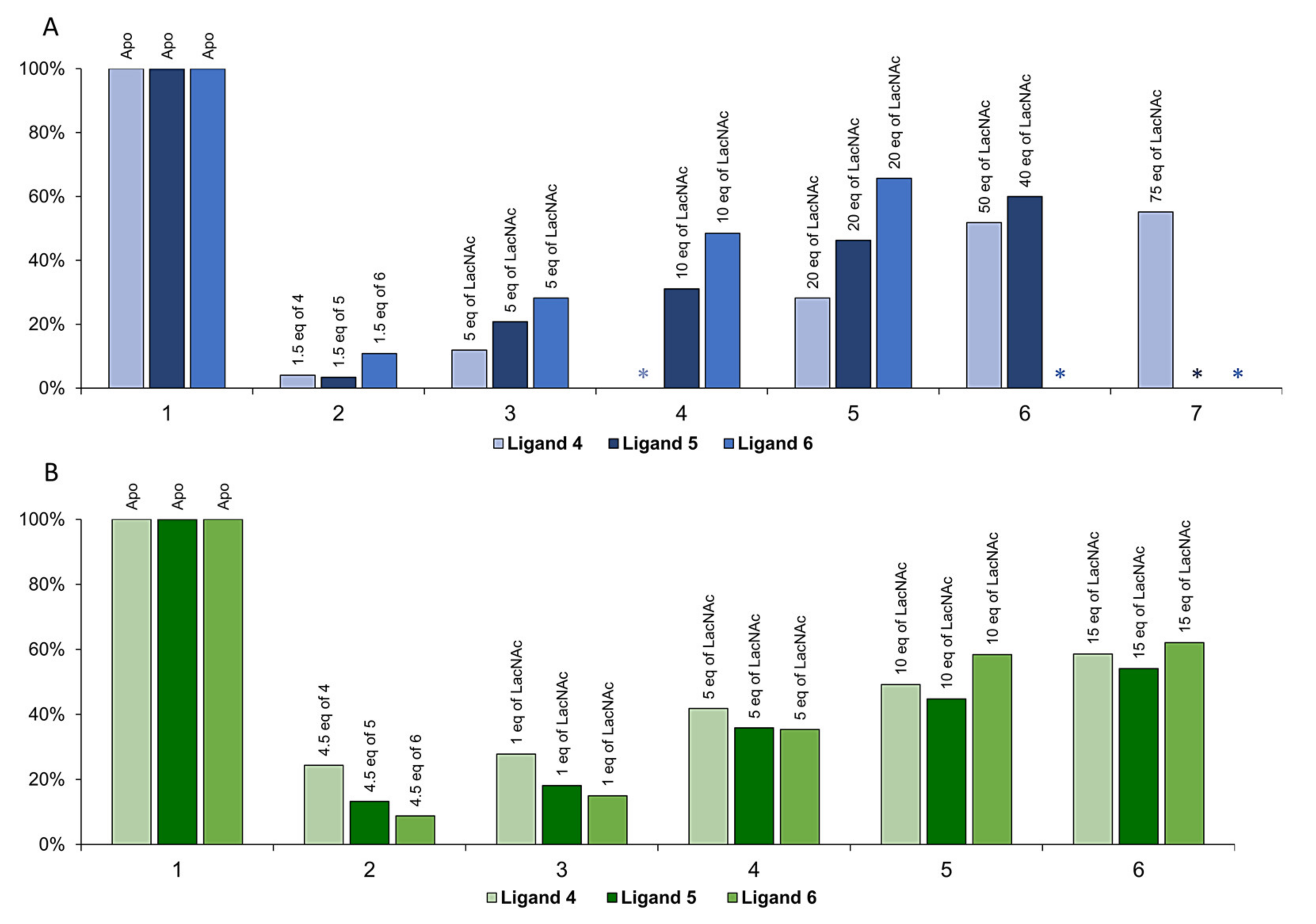
2.2. Inhibitory Activity Evaluation
2.3. Molecular Recognition
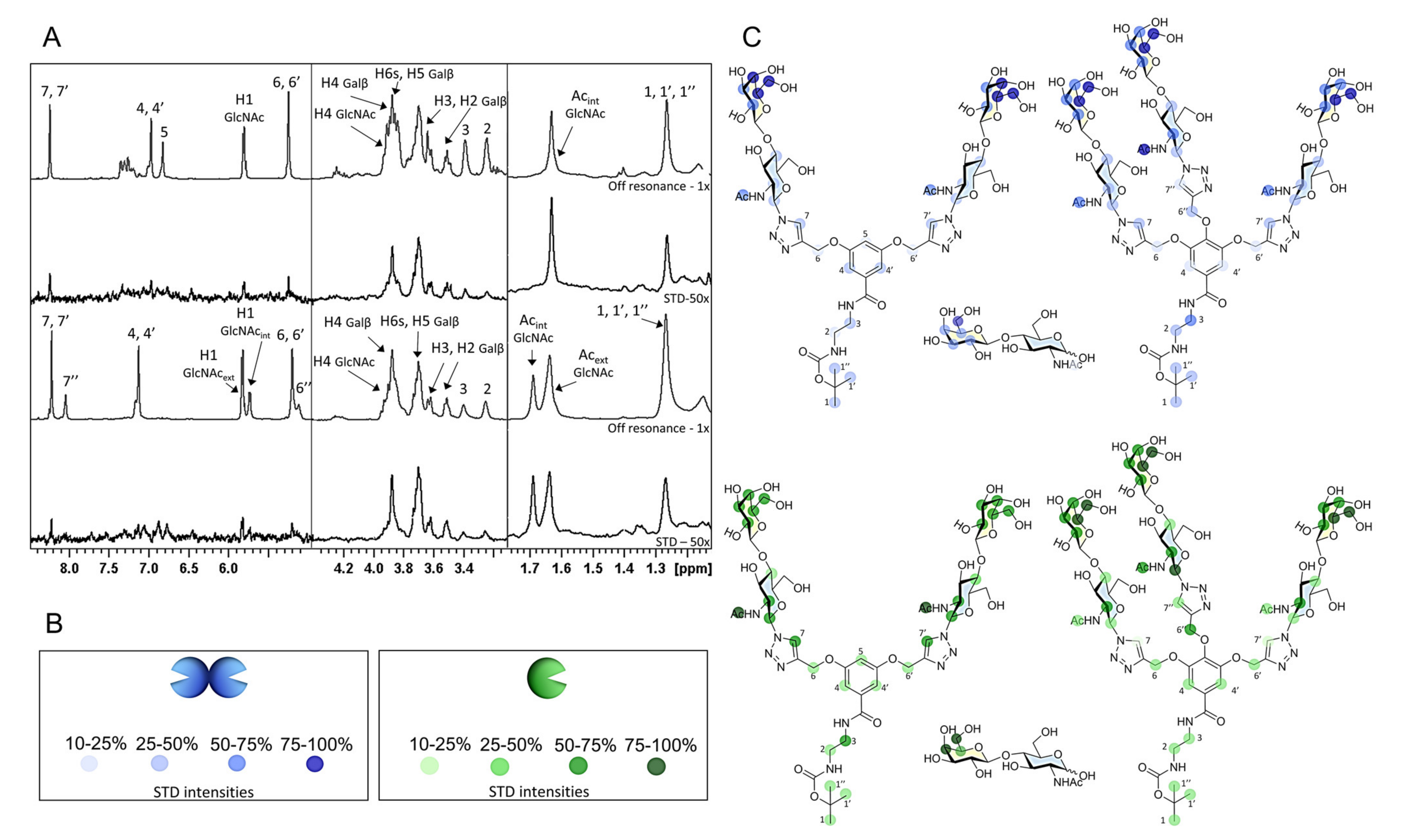
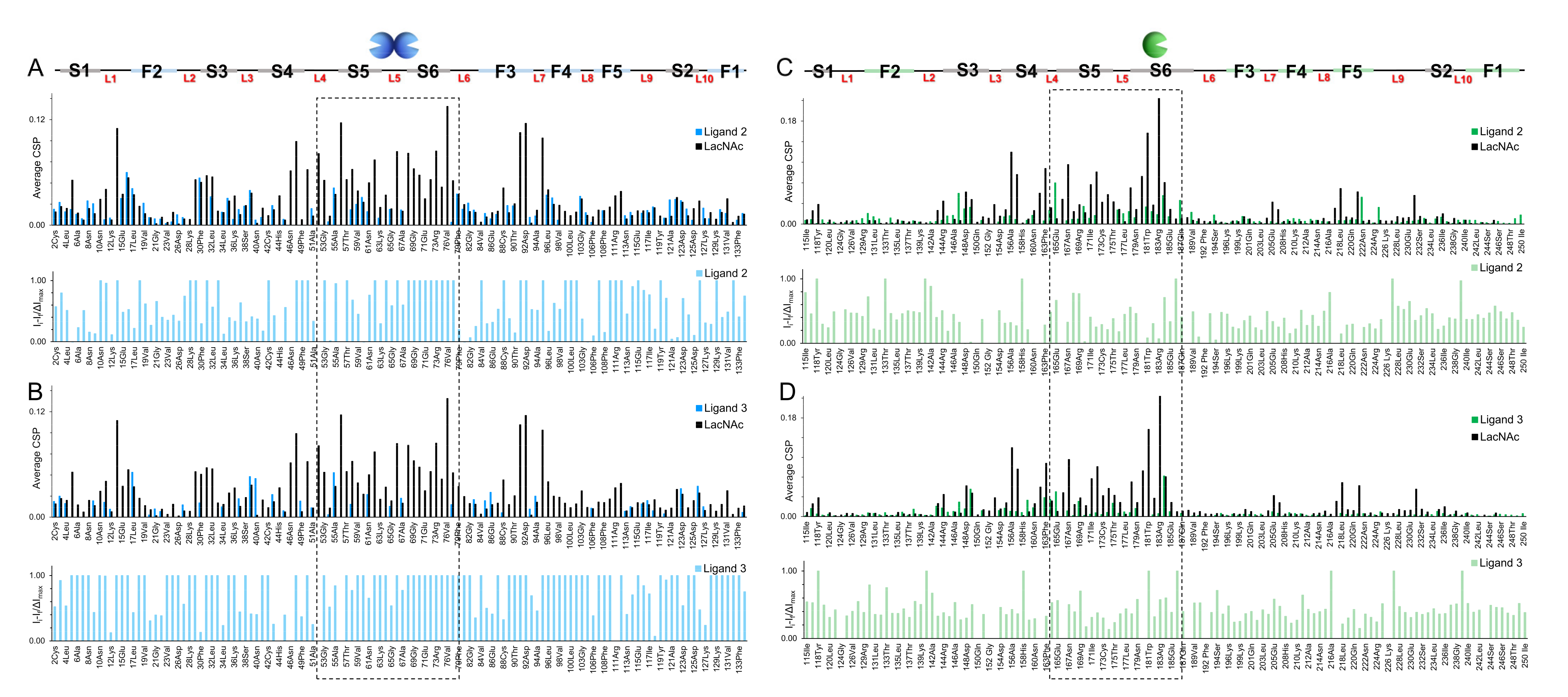
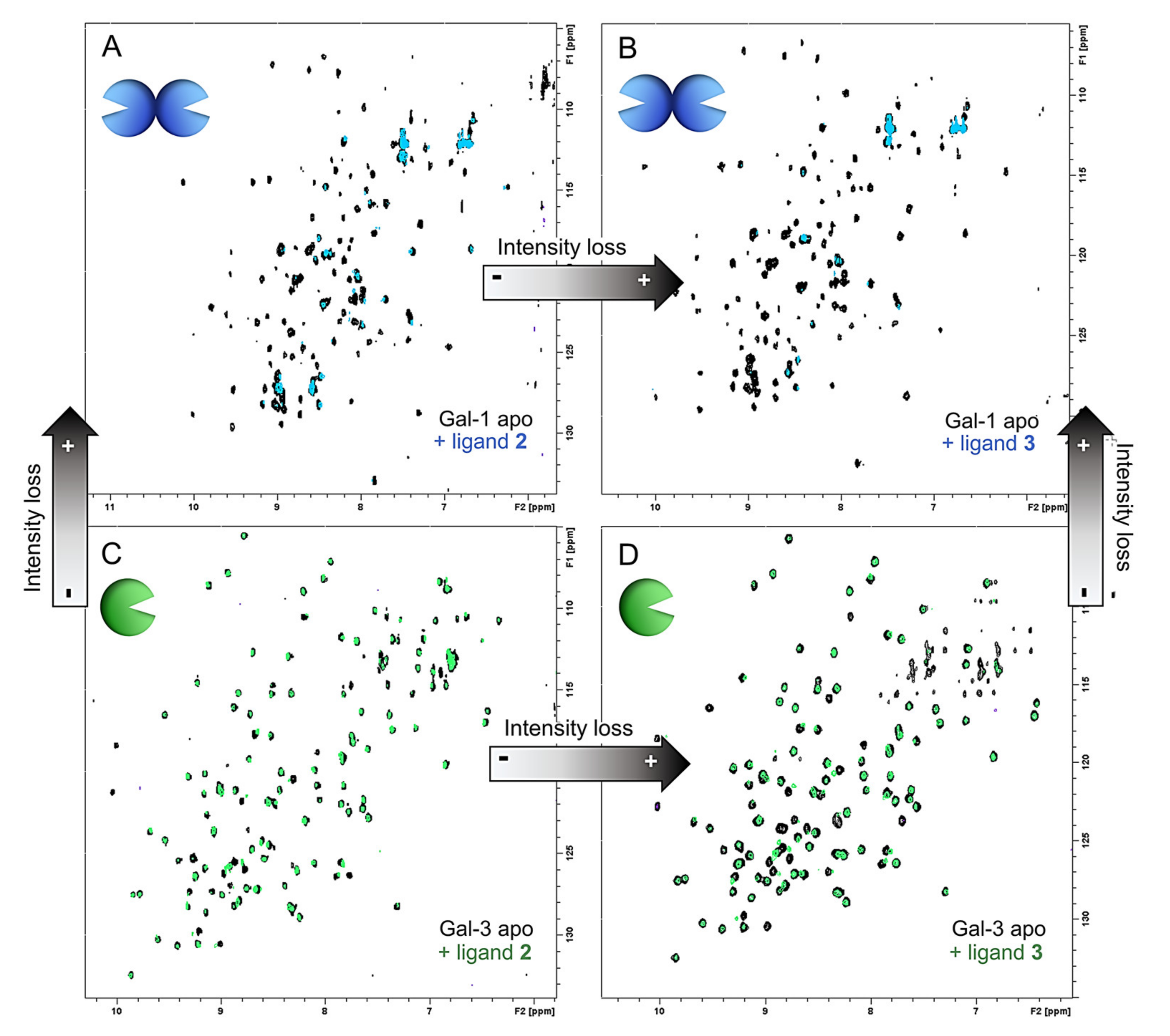
2.3.1. NMR Studies
2.3.2. DLS and Cryo-EM Measurements
3. Materials and Methods
3.1. Synthesis of Ligands 2–6
3.1.1. Synthesis of Ligands 2 and 3
3.1.2. Synthesis of Ligands 4–6
3.2. Size Exclusion Chromatography
3.3. Ultraviolet−Visible Spectrophotometry
3.4. Competitive ELISA Assay
3.5. Galectin Expression
3.5.1. Expression of Gal-1 and Gal-3 CRD Unlabeled
3.5.2. Expression of Gal-1 and Gal-3 CRD 15N Labeled
3.6. Galectin Purification
3.7. NMR Spectroscopy
3.7.1. Saturation Transfer Difference (STD) NMR
3.7.2. Chemical Shift Perturbation (CSP) Analysis
3.8. Cryo-EM Sample Preparation and Data Collection
3.9. DLS Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laaf, D.; Bojarová, P.; Elling, L.; Křen, V. Galectin–Carbohydrate Interactions in Biomedicine and Biotechnology. Trends Biotechnol. 2019, 37, 402–415. [Google Scholar] [CrossRef]
- Ebrahim, A.H.; Alalawi, Z.; Mirandola, L.; Rakhshanda, R.; Dahlbeck, S.; Nguyen, D.; Jenkins, M.; Grizzi, F.; Cobos, E.; Figueroa, J.A.; et al. Galectins in cancer: Carcinogenesis, diagnosis and therapy. Ann. Transl. Med. 2014, 2, 88. [Google Scholar] [CrossRef]
- Cousin, J.; Cloninger, M. The Role of Galectin-1 in Cancer Progression, and Synthetic Multivalent Systems for the Study of Galectin-1. Int. J. Mol. Sci. 2016, 17, 1566. [Google Scholar] [CrossRef] [Green Version]
- Nangia-Makker, P.; Balan, V.; Raz, A. Regulation of tumor progression by extracellular galectin-3. Cancer Microenviron. 2008, 1, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Dings, R.; Miller, M.; Griffin, R.; Mayo, K. Galectins as Molecular Targets for Therapeutic Intervention. Int. J. Mol. Sci. 2018, 19, 905. [Google Scholar] [CrossRef] [Green Version]
- Stillman, B.N.; Hsu, D.K.; Pang, M.; Brewer, C.F.; Johnson, P.; Liu, F.-T.; Baum, L.G. Galectin-3 and Galectin-1 Bind Distinct Cell Surface Glycoprotein Receptors to Induce T Cell Death. J. Immunol. 2006, 176, 778–789. [Google Scholar] [CrossRef] [Green Version]
- Girard, A.; Magnani, J.L. Clinical Trials and Applications of Galectin Antagonists. Trends Glycosci. Glycotechnol. 2018, 30, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Bertuzzi, S.; Quintana, J.I.; Ardá, A.; Gimeno, A.; Jiménez-Barbero, J. Targeting Galectins with Glycomimetics. Front. Chem. 2020, 8, 593. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Cumashi, A.; Bianco, G.A.; Ciavardelli, D.; Iurisci, I.; D’Egidio, M.; Piccolo, E.; Tinari, N.; Nifantiev, N.; Iacobelli, S. Synthetic lactulose amines: Novel class of anticancer agents that induce tumor-cell apoptosis and inhibit galectin-mediated homotypic cell aggregation and endothelial cell morphogenesis. Glycobiology 2006, 16, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Laaf, D.; Steffens, H.; Pelantová, H.; Bojarová, P.; Křen, V.; Elling, L. Chemo-Enzymatic Synthesis of Branched N-Acetyllactosamine Glycan Oligomers for Galectin-3 Inhibition. Adv. Synth. Catal. 2017, 359, 4015–4024. [Google Scholar] [CrossRef]
- Laaf, D.; Bojarová, P.; Pelantová, H.; Křen, V.; Elling, L. Tailored Multivalent Neo-Glycoproteins: Synthesis, Evaluation, and Application of a Library of Galectin-3-Binding Glycan Ligands. Bioconjug. Chem. 2017, 28, 2832–2840. [Google Scholar] [CrossRef]
- Zhang, H.; Laaf, D.; Elling, L.; Pieters, R.J. Thiodigalactoside-Bovine Serum Albumin Conjugates as High-Potency Inhibitors of Galectin-3: An Outstanding Example of Multivalent Presentation of Small Molecule Inhibitors. Bioconjug. Chem. 2018, 29, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Soomro, Z.H.; Cecioni, S.; Blanchard, H.; Praly, J.P.; Imberty, A.; Vidal, S.; Matthews, S.E. CuAAC synthesis of resorcin[4]arene-based glycoclusters as multivalent ligands of lectins. Org. Biomol. Chem. 2011, 9, 6587–6597. [Google Scholar] [CrossRef]
- Sakamoto, J.I.; Koyama, T.; Miyamoto, D.; Yingsakmongkon, S.; Hidari, K.I.P.J.; Jampangern, W.; Suzuki, T.; Suzuki, Y.; Esumi, Y.; Hatano, K.; et al. Thiosialoside clusters using carbosilane dendrimer core scaffolds as a new class of influenza neuraminidase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Nagahori, N.; Nishimura, S.-I. Direct and Efficient Monitoring of Glycosyltransferase Reactions on Gold Colloidal Nanoparticles by Using Mass Spectrometry. Chem. Eur. J. 2006, 12, 6478–6485. [Google Scholar] [CrossRef]
- Heine, V.; Hovorková, M.; Vlachová, M.; Filipová, M.; Bumba, L.; Janoušková, O.; Hubálek, M.; Cvačka, J.; Petrásková, L.; Pelantová, H.; et al. Immunoprotective neo-glycoproteins: Chemoenzymatic synthesis of multivalent glycomimetics for inhibition of cancer-related galectin-3. Eur. J. Med. Chem. 2021, 220, 113500. [Google Scholar] [CrossRef]
- Gimeno, A.; Delgado, S.; Valverde, P.; Bertuzzi, S.; Berbís, M.A.; Echavarren, J.; Lacetera, A.; Martín-Santamaría, S.; Surolia, A.; Cañada, F.J.; et al. Minimizing the Entropy Penalty for Ligand Binding: Lessons from the Molecular Recognition of the Histo Blood-Group Antigens by Human Galectin-3. Angew. Chem. Int. Ed. 2019, 58, 7268–7272. [Google Scholar] [CrossRef] [Green Version]
- Bertuzzi, S.; Gimeno, A.; Núñez-Franco, R.; Bernardo-Seisdedos, G.; Delgado, S.; Jiménez-Osés, G.; Millet, O.; Jiménez-Barbero, J.; Ardá, A. Unravelling the Time Scale of Conformational Plasticity and Allostery in Glycan Recognition by Human Galectin-1. Chem. Eur. J. 2020, 26, 15643–15653. [Google Scholar] [CrossRef]
- Ardá, A.; Jiménez-Barbero, J. The recognition of glycans by protein receptors. Insights from NMR spectroscopy. Chem. Commun. 2018, 54, 4761–4769. [Google Scholar] [CrossRef]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef]
- Chytil, P.; Koziolová, E.; Etrych, T.; Ulbrich, K. HPMA Copolymer-Drug Conjugates with Controlled Tumor-Specific Drug Release. Macromol. Biosci. 2018, 18, 1700209. [Google Scholar] [CrossRef]
- Filipová, M.; Bojarová, P.; Rodrigues Tavares, M.; Bumba, L.; Elling, L.; Chytil, P.; Gunár, K.; Křen, V.; Etrych, T.; Janoušková, O. Glycopolymers for Efficient Inhibition of Galectin-3: In Vitro Proof of Efficacy Using Suppression of T Lymphocyte Apoptosis and Tumor Cell Migration. Biomacromolecules 2020, 21, 3122–3133. [Google Scholar] [CrossRef]
- Bojarová, P.; Tavares, M.R.; Laaf, D.; Bumba, L.; Petrásková, L.; Konefał, R.; Bláhová, M.; Pelantová, H.; Elling, L.; Etrych, T.; et al. Biocompatible glyconanomaterials based on HPMA-copolymer for specific targeting of galectin-3. J. Nanobiotechnol. 2018, 16, 73. [Google Scholar] [CrossRef]
- Tavares, M.R.; Bláhová, M.; Sedláková, L.; Elling, L.; Pelantová, H.; Konefał, R.; Etrych, T.; Křen, V.; Bojarová, P.; Chytil, P. High-Affinity N-(2-Hydroxypropyl) methacrylamide Copolymers with Tailored N-Acetyllactosamine Presentation Discriminate between Galectins. Biomacromolecules 2020, 21, 641–652. [Google Scholar] [CrossRef]
- Bojarová, P.; Kulik, N.; Hovorková, M.; Slámová, K.; Pelantová, H.; Křen, V. The β-N-Acetylhexosaminidase in the Synthesis of Bioactive Glycans: Protein and Reaction Engineering. Molecules 2019, 24, 599. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, Z.; Nekvasilová, P.; Bojarová, P.; Křen, V.; Slámová, K. Advanced glycosidases as ingenious biosynthetic instruments. Biotechnol. Adv. 2021, 49, 107733. [Google Scholar] [CrossRef]
- Gimeno, A.; Valverde, P.; Ardá, A.; Jiménez-Barbero, J. Glycan structures and their interactions with proteins. A NMR view. Curr. Opin. Struct. Biol. 2020, 62, 22–30. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Meyer, B.; Peters, T. NMR Spectroscopy Techniques for Screening and Identifying Ligand Binding to Protein Receptors. Angew. Chem. Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-transfer difference (STD) NMR: A simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Šubr, V.; Ulbrich, K. Synthesis and properties of new N-(2-hydroxypropyl) methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React. Funct. Polym. 2006, 66, 1525–1538. [Google Scholar] [CrossRef]
- Chytil, P.; Etrych, T.; Kříž, J.; Šubr, V.; Ulbrich, K. N-(2-Hydroxypropyl) methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour targeting. Synthesis by RAFT polymerisation and physicochemical characterisation. Eur. J. Pharm. Sci. 2010, 41, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Laaf, D.; Spiwok, V.; Elling, L.; Křen, V.; Bojarová, P. Poly-N-Acetyllactosamine Neo-Glycoproteins as Nanomolar Ligands of Human Galectin-3: Binding Kinetics and Modeling. Int. J. Mol. Sci. 2018, 19, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | LacNAc Presentation | LacNAc Content [mol.%] b | Mn [g mol−1] c | Mw [g mol−1] c | Đ c |
|---|---|---|---|---|---|
| 4 | Individual | 19.3 | 25,200 | 33,600 | 1.3 |
| 5 | bivalent branching | 20.9 | 30,200 | 37,900 | 1.3 |
| 6 | trivalent branching | 22.0 | 32,400 | 38,400 | 1.2 |
| Ligand 1 | Ligand 2 | Ligand 3 | Ligand 4 | Ligand 5 | Ligand 6 | |
|---|---|---|---|---|---|---|
| Gal-1 | 78 ± 23 | 19 ± 5 | 9 ± 3 | 0.086 ± 0.05 | 0.41 ± 0.08 | 0.082 ± 0.02 |
| rp/n | 1 | 2 | 2.8 | 906 | 95 | 317 |
| Gal-3 | 44 ± 8 | 12 ± 2 | 4.4 ± 1.6 | 11 ± 1 | 4.6 ± 1.3 | 1.7 ± 0.4 |
| rp/n | 1 | 1.8 | 3.3 | 4 | 4.8 | 8.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertuzzi, S.; Gimeno, A.; Martinez-Castillo, A.; Lete, M.G.; Delgado, S.; Airoldi, C.; Rodrigues Tavares, M.; Bláhová, M.; Chytil, P.; Křen, V.; et al. Cross-Linking Effects Dictate the Preference of Galectins to Bind LacNAc-Decorated HPMA Copolymers. Int. J. Mol. Sci. 2021, 22, 6000. https://doi.org/10.3390/ijms22116000
Bertuzzi S, Gimeno A, Martinez-Castillo A, Lete MG, Delgado S, Airoldi C, Rodrigues Tavares M, Bláhová M, Chytil P, Křen V, et al. Cross-Linking Effects Dictate the Preference of Galectins to Bind LacNAc-Decorated HPMA Copolymers. International Journal of Molecular Sciences. 2021; 22(11):6000. https://doi.org/10.3390/ijms22116000
Chicago/Turabian StyleBertuzzi, Sara, Ana Gimeno, Ane Martinez-Castillo, Marta G. Lete, Sandra Delgado, Cristina Airoldi, Marina Rodrigues Tavares, Markéta Bláhová, Petr Chytil, Vladimír Křen, and et al. 2021. "Cross-Linking Effects Dictate the Preference of Galectins to Bind LacNAc-Decorated HPMA Copolymers" International Journal of Molecular Sciences 22, no. 11: 6000. https://doi.org/10.3390/ijms22116000