Monitoring the Site-Specific Solid-State NMR Data in Oligopeptides


Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovsky Square #2, 16206 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(8), 2700; https://doi.org/10.3390/ijms21082700
Submission received: 24 March 2020
/
Revised: 6 April 2020
/
Accepted: 9 April 2020
/
Published: 13 April 2020
(This article belongs to the Special Issue Molecular Structure and Dynamics Probed by Spectroscopic Techniques and Computational Approaches: New Trends by NMR, FTIR, Neutron Scattering and Simulation)
Abstract
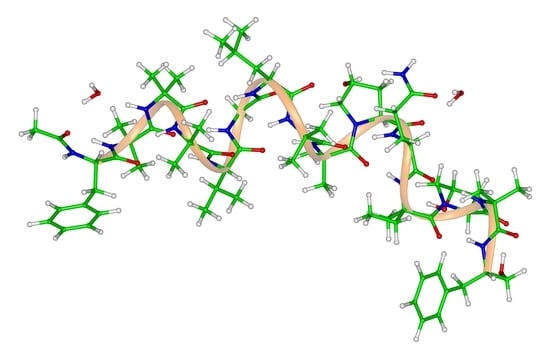
:Reliable values of the solid-state NMR (SSNMR) parameters together with precise structural data specific for a given amino acid site in an oligopeptide are needed for the proper interpretation of measurements aiming at an understanding of oligopeptides’ function. The periodic density functional theory (DFT)-based computations of geometries and SSNMR chemical shielding tensors (CSTs) of solids are shown to be accurate enough to support the SSNMR investigations of suitably chosen models of oriented samples of oligopeptides. This finding is based on a thorough comparison between the DFT and experimental data for a set of tripeptides with both 13Cα and 15Namid CSTs available from the single-crystal SSNMR measurements and covering the three most common secondary structural elements of polypeptides. Thus, the ground is laid for a quantitative description of local spectral parameters of crystalline oligopeptides, as demonstrated for the backbone 15Namid nuclei of samarosporin I, which is a pentadecapeptide (composed of five classical and ten nonproteinogenic amino acids) featuring a strong antimicrobial activity.
1. Introduction
Oligopeptides have been intensely studied due to their numerous applications [1]. One of the most important areas of the research aims at finding an alternative antimicrobial strategy [2]. Related host-defense oligopeptides [3], mostly of the peptaibol family [4], have been analyzed in terms of their function, which is based on an ability to form pores in biological membranes [5]. The solid-state nuclear magnetic resonance (SSNMR) measurements are crucial in understanding the pore formation properties of peptaibols [6]. Due to the large size and complexity of peptaibols and similar oligopeptides, their SSNMR investigations usually do not use the site-specific spectroscopical/structural data for every investigated peptide unit. Instead, either of the two strategies is adopted. In the first approach, “collective” values for a group of nuclei are employed (for example, for the 15N chemical shift tensors (CSTs) of amidic nitrogens in classical amino acid and α-amino isobutyric acid (Aib) residues, thus neglecting differences of CSTs within each group [7]). In the second approach, site-specific results are obtained only for some atoms, which are isotope-labeled (see impressive efforts by the group of Naito to study eleven alamethicin molecules singly labeled with 13C at the position of respective carbonyl carbon [8]), or chemically introduced in case of 19F [9]. Importantly, the plane wave density functional theory (PW DFT)-based calculations of periodic structures and their NMR properties [10] can currently be applied even to bigger oligopeptides of up to about 20 amino acids (depending on the number of formula units in the crystal’s unit cell), and may provide useful information about differences in the local geometry and SSNMR parameters within, for instance, the whole backbone of a peptaibol [11]. It is thus of interest to calibrate the results of the PW DFT computations against highly accurate measurements, such as those described below, which were taken from single crystal studies [12,13,14], in order to establish if it could be possible to reliably predict a site-specific variation in the SSNMR data and include this information in analyses of experiments on larger oligopeptides [15]. This type of monitoring is presented here for samarosporin I (a naturally occurring peptaibol comprised of 15 amino acids [16]) on the basis of benchmarking calculations for a set of six triglycines, and for N-Ac-Aib-OH, N-Ac-Leu-OH and Ala-Pro-Gly dihydrate, after their assessment performed for melanostatin (Pro-Leu-Gly-NH2 hemihydrate [17]). The results directly capture an influence of secondary structural elements upon the NMR parameters (see reference [18] for the most recent review of this topic) and could be important in NMR crystallography [19,20,21,22,23] of oligopeptides and in an interpretation of spectra of their oriented samples [24].
2. Results
2.1. The Chemical Shielding Tensors of Triglycines
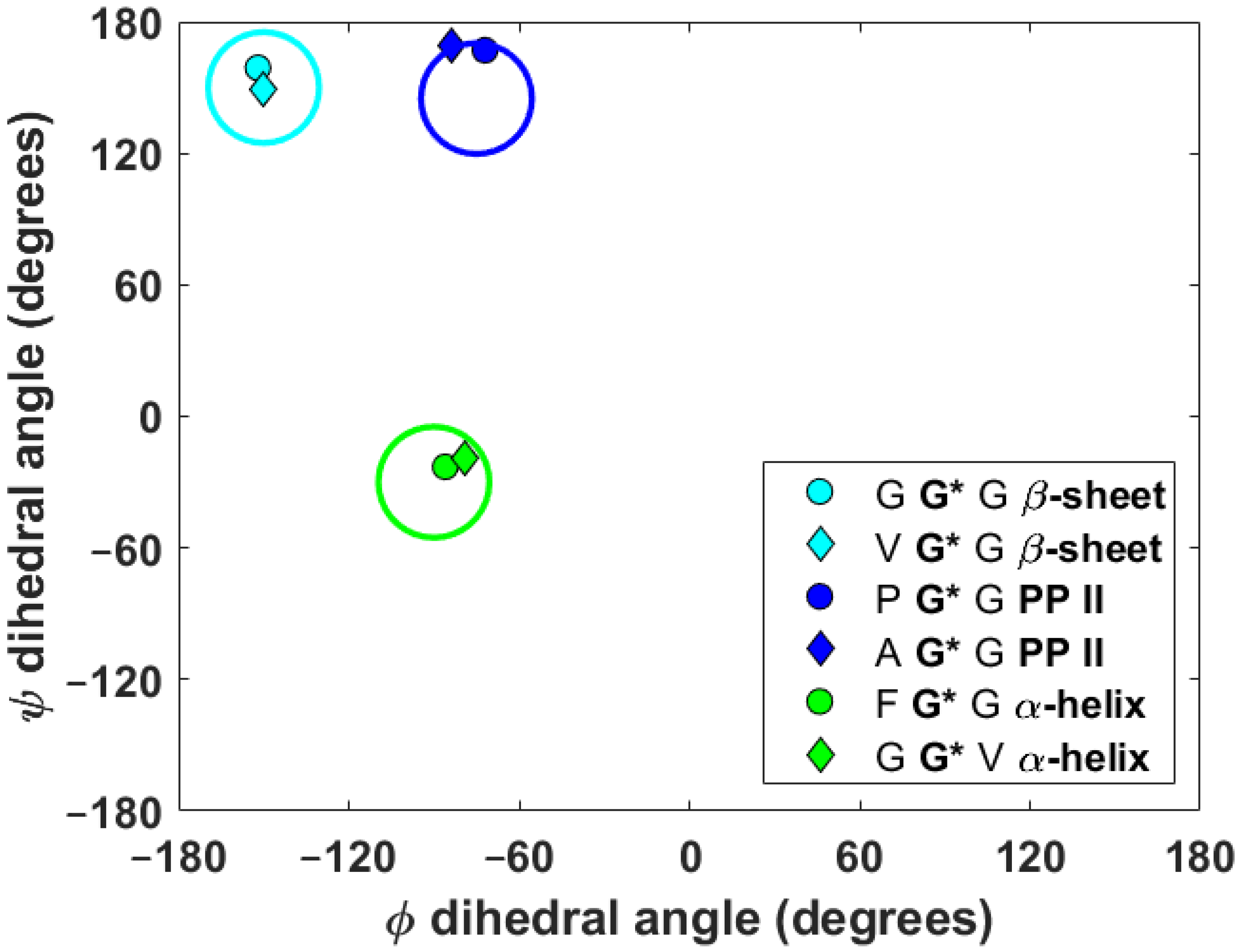
Some time ago, Wittebort et al. performed meticulous single-crystal (SC) SSNMR measurements of central glycyl 13Cα and 15Namid CSTs in two relatively large series of tripeptides of known solid-phase geometry, featuring the torsion angles typical for common secondary structural motifs found in polypeptides [12,13,14]. Here, six tripeptides (specified in Table 1 using their simplified names in apostrophes) are taken from the sets studied by Wittebort et al. so that both the 13Cα and 15Namid CSTs are available for two representatives of each of, arguably, the most frequent regular secondary structures: β-sheet, αR-helix, and the polyproline II (PP II) helix [25]. Figure 1 presents the Ramachandran plot of the {φ, ψ} angles obtained from the PW DFT optimization (see Part 4) of these six tripeptides.
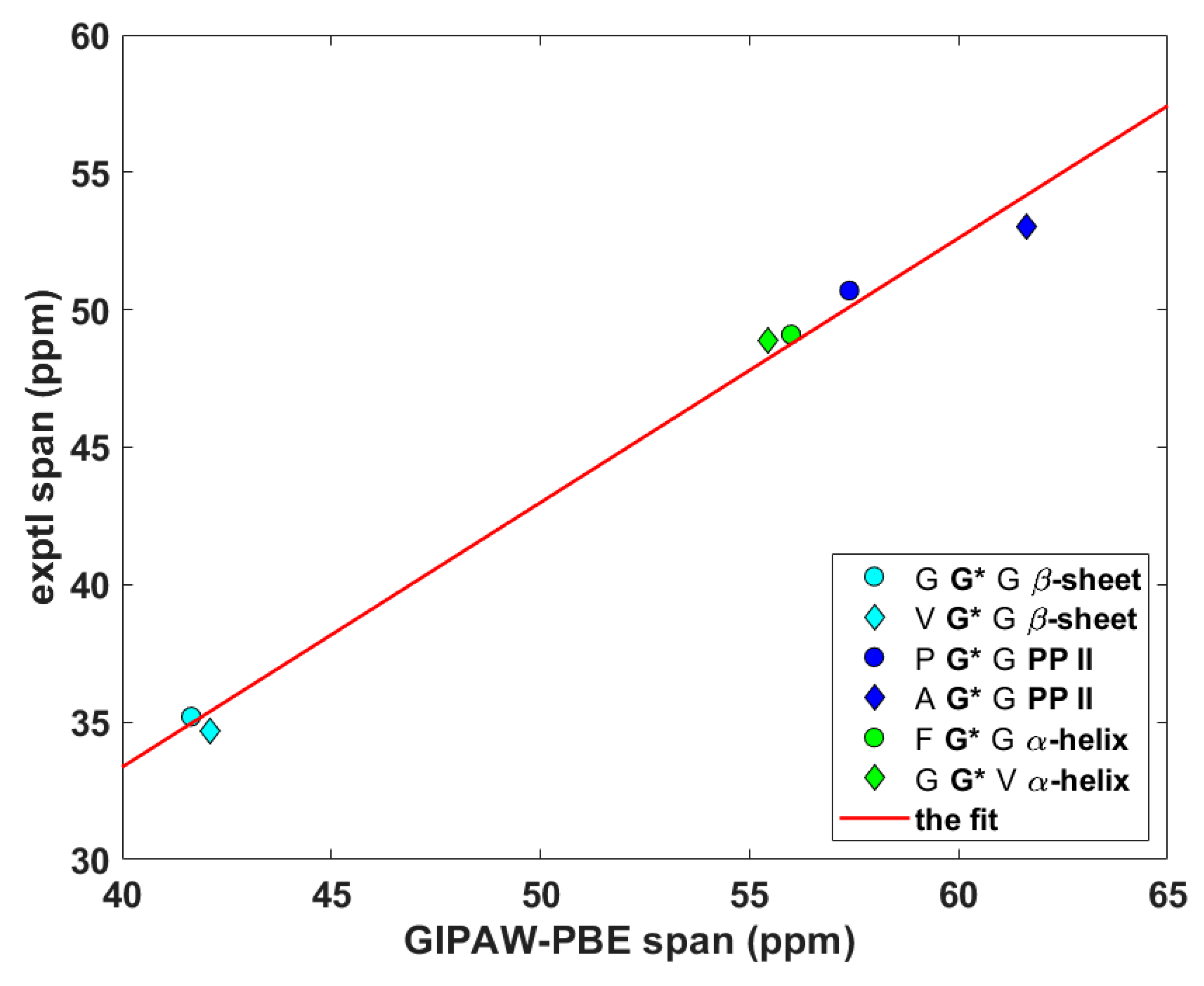
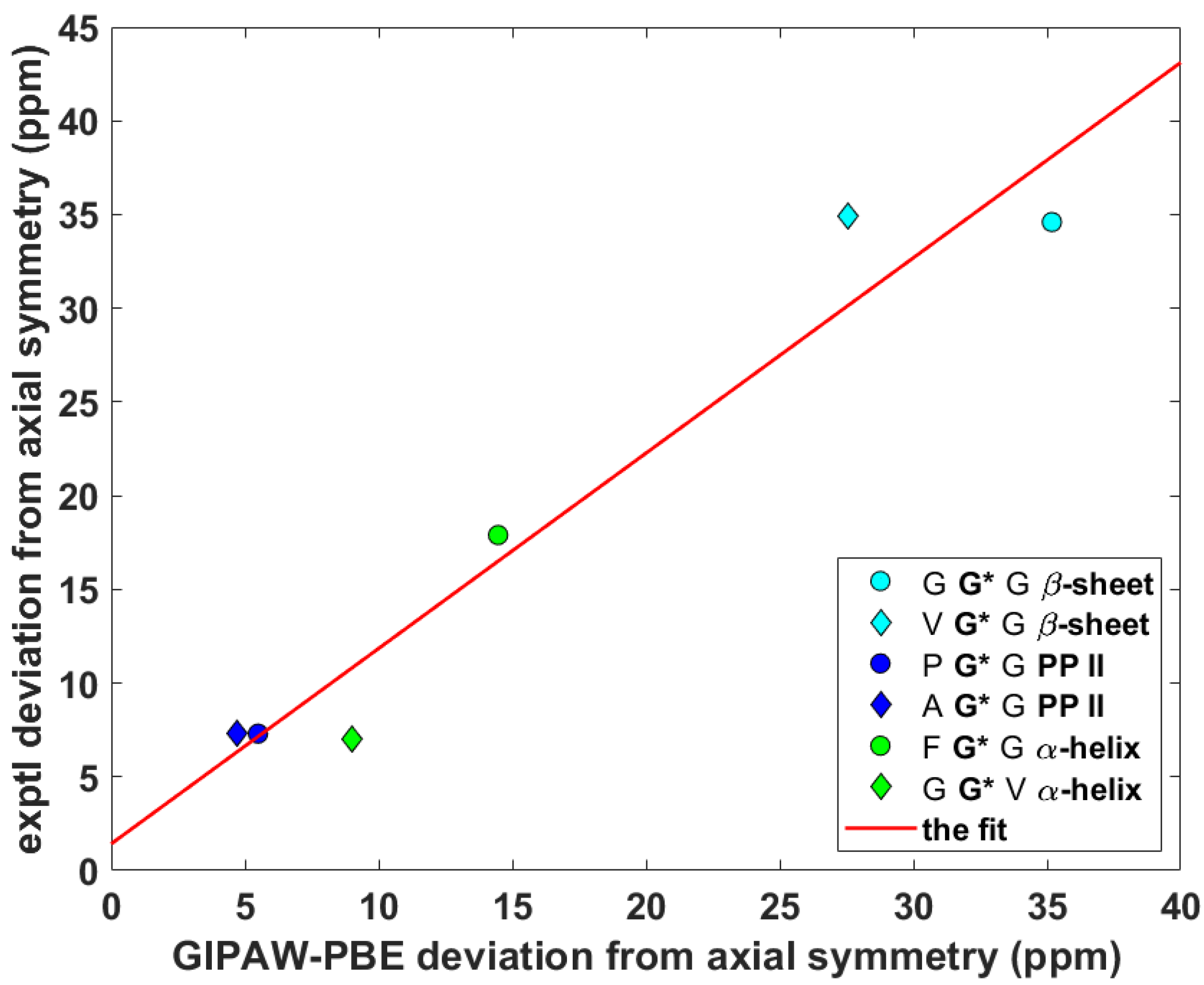
Wittebort et al. [12,13] discerned three important trends in the dependence of SC SSNMR parameters upon the secondary structure. First, they found that the 13Cα isotropic chemical shifts, δiso, of glycyls were always lower in α-helices than in β-sheets and PP II helices. Second, values of the span, δspan, of 13Cα CSTs, δspan = δ33 – δ11 for δ11 ≤ δ22 ≤ δ33 ordering of the eigenvalues of the chemical shift tensor, were the highest for glycyls in PP II, followed by those in α-helices and then by those in β-sheets. Third, values of the deviation from the axial symmetry, δdev, of 15Namid CSTs, δdev = δ22 – δ11 for the same ordering as above, were the highest for glycyls in β-sheets, followed by those in PP II and then by those in α-helices. Thus, an unambiguous identification of the secondary structure of a peptide should be possible by combining the three experimental trends [13]. Here, the ability of the gauge-including projector augmented wave (GIPAW) Perdew–Burke–Ernzerhof (PBE) calculations (see Materials and Methods section) of the chemical shielding tensors to reproduce the aforementioned tendencies is tested. The eigenvalues, both theoretical and experimental, obtained for the six tripeptides are summarized in Supporting Materials Tables S1 (15Namid) and S2 (13Cα). The 13Cα δiso and δspan, and 15Namid δdev SSNMR parameters are graphically shown in Figure 2, Figure 3 and Figure 4 (fits of the eigenvalues are included in Supporting Materials). Expectedly [26], these figures illustrate a full agreement between the measured dependences and their theoretical counterparts, which were obtained from the GIPAW PBE calculations of the chemical shielding after the PW PBE optimizations of crystalline peptides’ geometries.
2.2. The Calibration of the 15N Chemical Shielding for Peptides
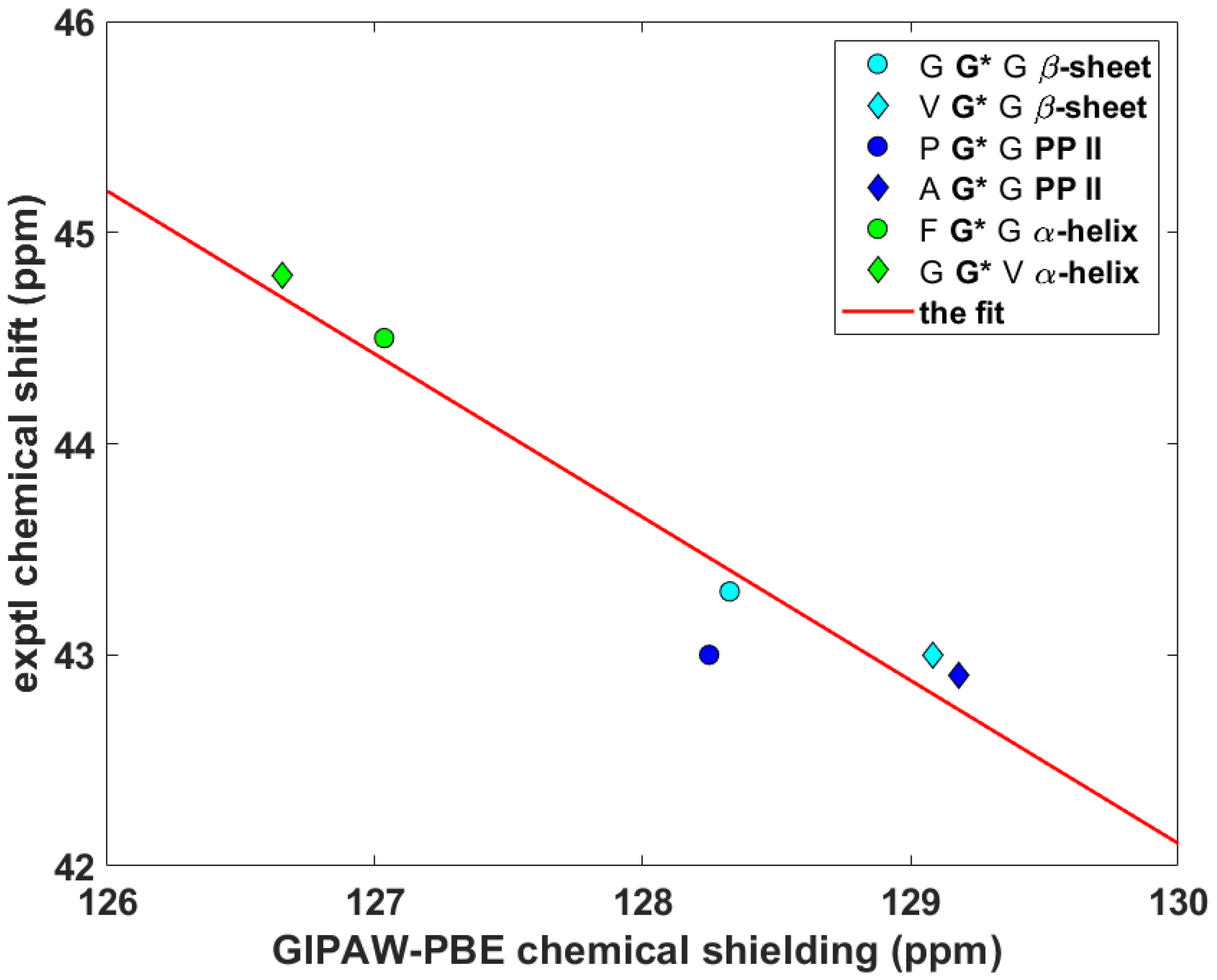
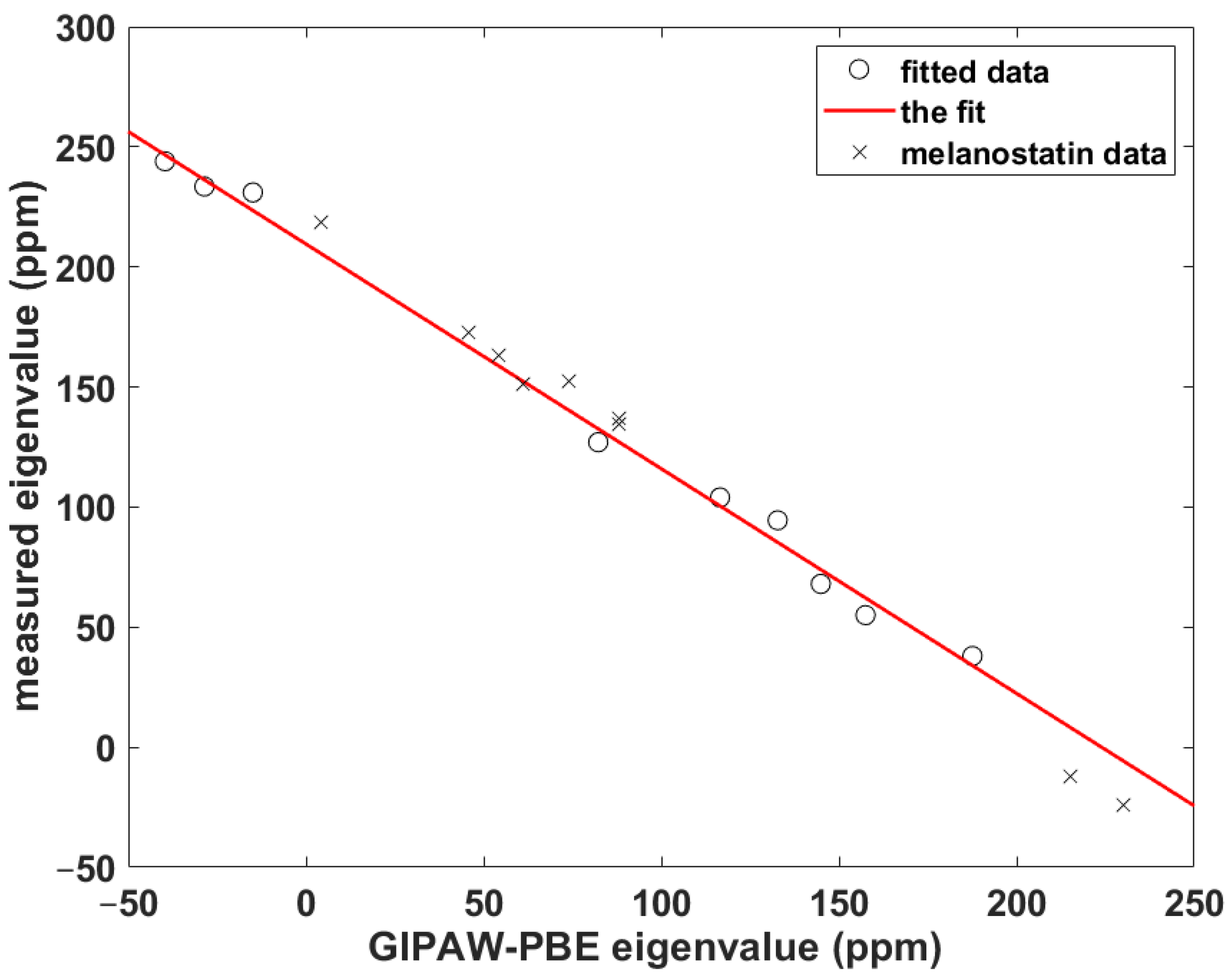
For cases when the experimental information is incomplete or currently unavailable, it is crucial to be able to quantitatively predict the parameters of the 15Namid chemical shift tensor, namely, its eigenvalues and their orientation in the molecular frame, for their further use in simulations of the SSNMR spectra of peptaibols (see the review [27] and references cited therein). Importantly, an unbiased calibration of the relationship between the chemical shift and computed chemical shielding data needs to include some non-canonical amino acid(s), because peptaibols contain quaternary residues such as l-4-hydroxyproline (Hyp), d-isovaline (Iva) or the aforementioned Aib, while experimental studies of peptides containing those residues are scarce [7]. As a consequence, the benchmark set consists of just three systems: N-Ac-Aib-OH and N-Ac-Leu-OH from the SSNMR study of powders [7] and the prolyl data of Ala-Pro-Gly dihydrate obtained by Wittebort et al. [14]. The ability to accurately predict the 15Namid chemical shift tensor components using this benchmark set is verified for Pro, Leu and Gly sites of melanostatin (it is noted that in reference [17] the experimental data were reported in nitromethane scale [28] and in the icosahedral representation [29]). The computational procedure is based on our previous work [30], and it should be realized that it does not explicitly use a chemical shielding of any referencing species. It involves fitting of a set of δii to the corresponding set of σii to obtain the slope, a, and the intercept, b, in {σii} = a*{δii} + b, as illustrated in Figure 5 (in this shorthand notation, curly brackets indicate correctly ordered elements of both sets; see also reference [31]). Subsequently, the theoretical chemical shift, εiso, of a given nucleus is estimated from εiso = (ε11 + ε22 + ε33)/3, where εii = a*σii + b. In the present case, a = –0.93574, b = 209.54 ppm, adjusted R2 = 0.99359, one standard deviation = 6.1 ppm (the underlying data are gathered in Table S1). As follows from an inspection of Table 2, the differences are minimal (smaller than one ppm) between the measured and theoretical isotropic chemical shifts of the amidic nitrogens in melanostatin. Hence, this calibration is used to estimate the 15Namid chemical shift data of samarosporin I that are discussed below.
Regarding an orientation of the CSTs in the molecular/crystal frame, we recently showed the GIPAW PBE calculations to provide highly accurate results for Nδ and Nε sites in l-histidine hydrochloride monohydrate [32]. Nevertheless, the SC SSNMR measurements by Wittebort et al. [14] of the central glycyl 15Namid sites in AGG and GGV and of the prolyl 15Namid in APG (see Table 1) are employed here to confirm the quality of the PW DFT predictions. The experimental values of the set of angles from reference [14] are shown in Table 3 together with the corresponding theoretical results (reconstructed using the specific peptide plane definitions given below Table 2 of reference [14]), which can be seen to be quantitatively correct.
2.3. Predictions for Samarosporin I
Samarosporin I is the subfamily 2 peptaibol [33]. Its sequence is Ac-Phe-Aib-Aib-Aib-Val-Gly-Leu-Aib-Aib-Hyp-Gln-Iva-Hyp-Aib-Fol (Aib, Hyp, and Iva were defined above, while Ac and Fol, respectively, denote acetyl and phenylalaninol at the N-end and C-end of the peptide). Samarosporin I, due to its strong antimicrobial activity [34], was studied by SC X-ray diffraction (XRD) at two temperatures (100 and 293 K) in order to help elucidate its mechanism of action [35]. An SSNMR investigation of this oligopeptide is clearly desirable and would benefit from the monitoring of the CSTs if, for instance, the so-called polarity index slant angle (PISA) wheels [36] were to be analyzed (see reference [37] for a survey of related experimental techniques). Specifically, the solid-phase structure of samarosporin I features right-handed helical folding. The helix tilt angle could thus be determined in oriented lipid bilayers from two-dimensional spectra correlating 1Hamid–15Namid dipolar couplings and 15Namid chemical shift anisotropy of atoms in peptide planes in a α-helical configuration (for details, see reference [37]). The relevant structural information about the samarosporin I backbone is collected in Table 4, namely, the values obtained after the PW PBE optimization of the Namid–Hamid bond lengths, rNH, and the {φ, ψ} dihedral angles. The {φ, ψ} values reported in Table 3 of reference [16] are also shown in Table 4 and indicate a close agreement between the DFT optimized and SC XRD structures of this oligopeptide. Additional structural information can be gleaned directly from the coordinates included as a PDB file in the Supporting Materials. The 15Namid CSTs are characterized in terms of the following parameters: an estimate, εiso, of the isotropic chemical shift; estimates, {ε11, ε22, ε33}, of principal components of the chemical shift tensor; the angles, {α, β, γ}, which describe an orientation of the CST in the crystal frame and are defined in Part 4 (numbering of atoms belonging to the respective reference planes is provided in Table S3). These results are further discussed in the next section.
3. Discussion
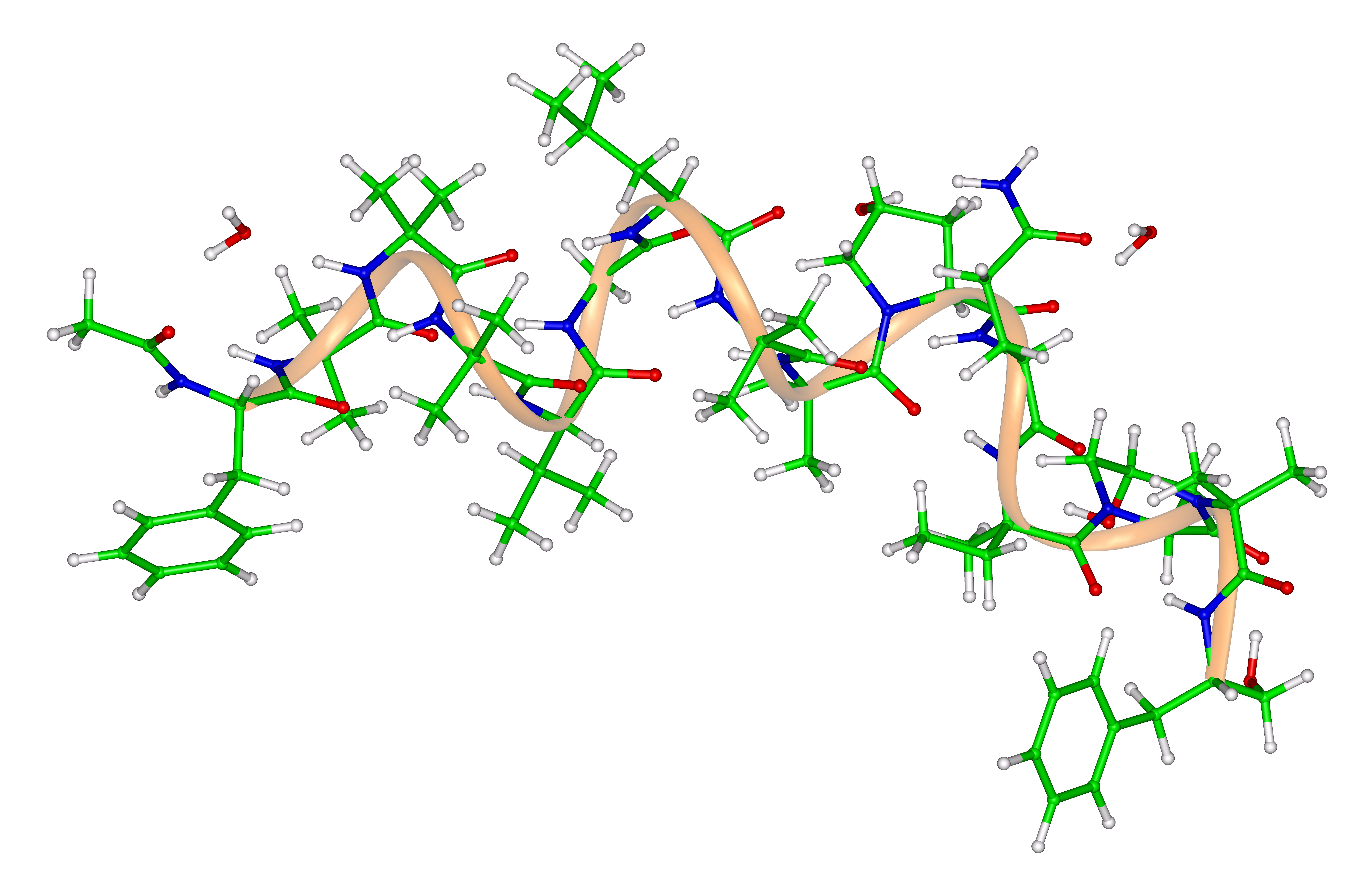
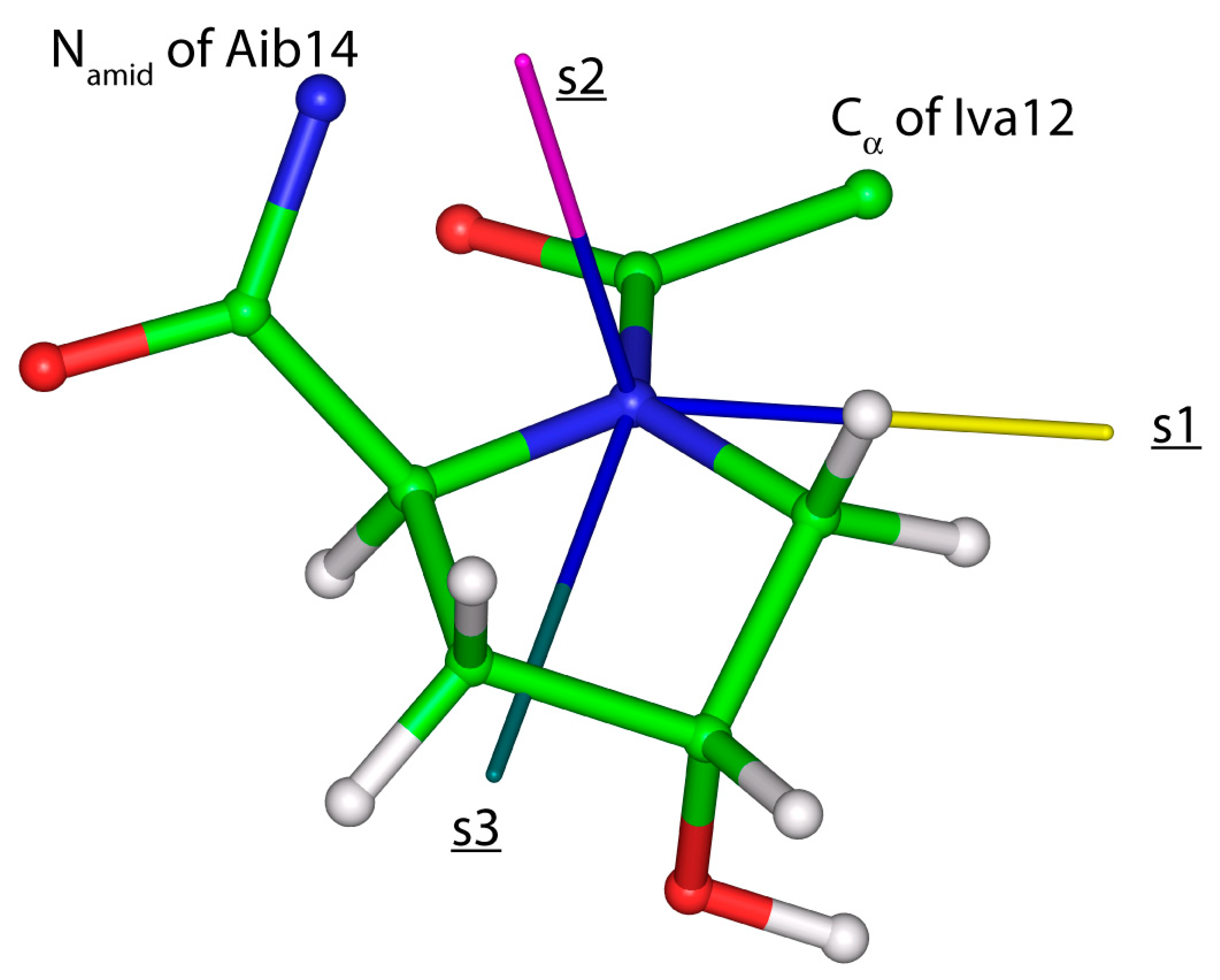
High-accuracy data are needed for establishment of a quantitatively accurate relationship between the structure of oligopeptides and their SSNMR parameters. Here, the data are carefully chosen from the results of the {13C, 15N} SC measurements previously performed for the central (13Cα and 15Namid) glycyl nuclei in tripeptides with known crystal geometries belonging to any of the three most common secondary structural elements found in proteins. This set of structures is employed to investigate the predictive power of the PW DFT computations, which is found to be strong for the key SSNMR parameters (13Cα δiso and δspan, and 15Namid δdev). In addition, orientations of the 15Namid CSTs are verified to be reliably described by the GIPAW PBE calculations carried out for the optimized geometries of AGG, GGV, and PGG. Hence, the predictive power of the PW DFT is used to calibrate the dependence of the chemical shift upon the computed chemical shielding (detailed in Section 2.2) and subsequently monitor the eigenvalues and orientations of the 15Namid CSTs along the backbone of an antimicrobial peptide samarosporin I. Importantly, the computations showed the same trend as was discerned experimentally for other peptaibols [38], namely, the 15Namid isotropic chemical shifts in Aib are significantly (by ca. 15 ppm in the present case) higher than in classical residues. An inspection of the averaged {ε11, ε22, ε33} values for these two groups of amino acids reveals the differences of the respective principal components leading to the distinctive isotropic chemical shifts. Specifically, the (“Aib”–“canonical”) differences, rounded to one ppm accordingly, amount to 14, 12, and 20 ppm for the ε11, ε22, and ε33 principal components. As for an orientation in the crystal frame of the 15Namid CSTs, given by a {α, β, γ} triple of angles, it should be mentioned that the most important are the values of β. This is because they directly enter a simulation of the PISA wheels [35] (and the values of α are in a typical case assumed to be zero). In the present case, both β and α angle values are, in general, similar to those found in a computational study of another oligopeptide, ampullosporin A [11]. Namely, β angles are slightly higher in proteinogenic amino acids (median of 20.1°) than in Aib (median of 15.3°) sites of samarosporin I, and α angles are negligibly small, with values below 5°. It is also worth mentioning that the estimated 15Namid chemical shift tensor components in hydroxyproline residues (Hyp10 and Hyp13) are close to the values found for nonproteinogenic amino acids (see Table 4), and hydroxyproline sites feature a specific orientation of their 15Namid CST. In particular, the eigenvector associated with the least shielded eigenvalue is significantly (by more than 20°) tilted off the peptide plane (see Figure 6). This information could be useful in future studies of prolyl-containing peptides [39].
4. Materials and Methods
A computational approach was adopted that applies Kohn–Sham DFT in the pseudopotential PW scheme and imposes periodic boundary conditions to treat the investigated crystal structure as an infinite system, as detailed in references [40,41,42]. First, the starting geometries specified in Table 1 were subjected to the optimization of all atoms’ positions with respect to the crystal lattice energy approximated with the Perdew–Burke–Ernzerhof (PBE) [43] exchange-correlation functional, while the unit-cell parameters were fixed at their corresponding XRD values. Subsequently, the CSTs were predicted employing the gauge-including projector augmented wave (GIPAW) [44,45] method combined with the PBE functional. The CASTEP 6.1 suite of codes [42] was used with the pseudopotentials generated on-the-fly, and with the thresholds and settings consistent with the “fine” level of accuracy of Materials Studio 5.0 software (the technical assistance was provided by Dr. M. Hušák, University of Chemistry and Technology, Prague, The Czech Republic. In particular, the cut-off energy of the plane-waves was set to 550 eV in all the above-mentioned calculations.
The optimized crystal geometries and the eigenvectors associated with respective eigenvalues were adopted to establish an orientation of the investigated 15Namid CSTs in the frame of the peptide bond. The reference plane was defined by the positions of Namid, Hamid, and Cα atoms of the given amino acid and using Namid–Hamid and Namid–Cα bond vectors. Then, the angle α is defined by a projection onto this plane of the eigenvector ξ1 associated with the most shielded eigenvalue, σ11; the angle β is subtended between the ξ1 and the related Namid–Hamid bond vector; and γ is the angle between a normal to the Namid; Hamid; Cα plane and the eigenvector ξ2 associated with the mid-shielded eigenvalue, σ22, of the 15Namid chemical shielding tensor in question. The eigenvectors were processed by the INFOR software [46] for their visualization in a crystal/molecular frame.
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/8/2700/s1.
Author Contributions
Conceptualization, J.C. and J.B.; Investigation, Writing, J.C.; Validation, Funding Acquisition, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the Ministry of Education, Youth and Sports of CR within the National Sustainability Program I, Project LO1507 POLYMAT.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hamley, I.W. Small Bioactive Peptides for Biomaterials Design and Therapeutics. Chem. Rev. 2017, 117, 14015–14041. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef]
- Yount, N.Y.; Weaver, D.C.; Lee, E.Y.; Lee, M.W.; Wang, H.; Chan, L.C.; Wong, G.C.L.; Yeaman, M.R. Unifying structural signature of eukaryotic α-helical host defense peptides. Proc. Natl. Acad. Sci. USA 2019, 116, 6944–6953. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Risinger, A.L.; Mitchell, C.A.; You, J.; Stamps, B.W.; Pan, N.; King, J.B.; Bopassa, J.C.; Judge, S.I.V.; Yang, Z.; et al. Unique amalgamation of primary and secondary structural elements transform peptaibols into potent bioactive cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2017, 114, 8957–8966. [Google Scholar] [CrossRef] [Green Version]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides with Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molugu, T.R.; Lee, S.; Brown, M.F. Concepts and Methods of Solid-State NMR Spectroscopy Applied to Biomembranes. Chem. Rev. 2017, 117, 12087–12132. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.; Bertani, P.; Raap, J.; Bechinger, B. Analysis of the amide 15N chemical shift tensor of the Cα tetrasubstituted constituent of membrane-active peptaibols, the α-aminoisobutyric acid residue, compared to those of di- and tri-substituted proteinogenic amino acid residues. J. Biomol. NMR 2009, 45, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Mishima, D.; Javkhlantugs, N.; Wang, J.; Ishioka, D.; Yokota, K.; Norisada, K.; Kawamura, I.; Ueda, K.; Naito, A. Structure and orientation of antibiotic peptide alamethicin in phospholipid bilayers as revealed by chemical shift oscillation analysis of solid state nuclear magnetic resonance and molecular dynamics simulation. BBA Biomembr. 2015, 1848, 2789–2798. [Google Scholar] [CrossRef] [Green Version]
- Grage, S.L.; Kara, S.; Bordessa, A.; Doan, V.; Rizzolo, F.; Putzu, M.; Kubař, T.; Papini, A.M.; Chaume, G.; Brigaud, T.; et al. Orthogonal 19F-Labeling for Solid-State NMR Spectroscopy Reveals the Conformation and Orientation of Short Peptaibols in Membranes. Chem. Eur. J. 2018, 24, 4238–4335. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azais, T.; Asbrook, S.E.; Griffin, J.M.; Yates, J.R.; Pickard, J.C. First-Principles Calculation of NMR Parameters Using the Gauge Including Projector Augmented Wave Method: A Chemist’s Point of View. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Theoretical Investigations Into the Variability of the N-15 Solid-State NMR Parameters Within an Antimicrobial Peptide Ampullosporin A. Phys. Res. 2018, 67, S349–S356. [Google Scholar] [CrossRef]
- Chekmenev, E.Y.; Xu, R.Z.; Mashuta, M.S.; Wittebort, R.J. Glycyl C alpha chemical shielding in tripeptides: Measurement by solid-state NMR and correlation with X-ray structure and theory. J. Am. Chem. Soc. 2002, 124, 11894–11899. [Google Scholar] [CrossRef]
- Chekmenev, E.Y.; Zhang, Q.W.; Waddell, K.W.; Mashuta, M.S.; Wittebort, R.J. N-15 chemical shielding in glycyl tripeptides: Measurement by solid-state NMR and correlation with x-ray structure. J. Am. Chem. Soc. 2004, 126, 379–384. [Google Scholar] [CrossRef]
- Waddell, K.W.; Chekmenev, E.Y.; Wittebort, R.J. Single-Crystal Studies of Peptide Prolyl and Glycyl 15N Shielding Tensors. J. Am. Chem. Soc. 2005, 127, 9030–9035. [Google Scholar] [CrossRef]
- Kang, X.; Elso, C.; Penfield, J.; Kirui, A.; Chen, A.; Zhang, L.; Wang, T. Integrated solid-state NMR and molecular dynamics modeling determines membrane insertion of human β-defensin analog. Commun. Biol. 2019, 2, 402–410. [Google Scholar] [CrossRef]
- Gessmann, R.; Axford, D.; Evans, G.; Bruckner, H.; Petratos, K. The crystal structure of samarosporin I at atomic resolution. J. Pept. Sci. 2012, 18, 678–684. [Google Scholar] [CrossRef]
- Strohmeier, M.; Grant, D.M. Experimental and theoretical investigation of the C-13 and N-15 chemical shift tensors in melanostatin-exploring the chemical shift tensor as a structural probe. J. Am. Chem. Soc. 2004, 126, 966–977. [Google Scholar] [CrossRef]
- Nerli, S.; McShan, A.C.; Sgourakis, N.G. Chemical shift-based methods in NMR structure determination. Prog. Nucl. Mag. Res. Sp. 2018, 106, 1–25. [Google Scholar] [CrossRef]
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. (Eds.) NMR Crystallography; Wiley: Chichester, UK, 2009. [Google Scholar]
- Hofstetter, A.; Balodis, M.; Paruzzo, F.M.; Widdifield, C.M.; Stevanato, G.; Pinon, A.C.; Bygrave, P.J.; Day, G.M.; Emsley, L. Rapid structure determination of molecular solids using chemical shifts directed by unambiguous prior constraints. J. Am. Chem. Soc. 2019, 141, 16624–16634. [Google Scholar] [CrossRef]
- Guzmán-Afonso, C.; Hong, Y.; Colaux, H.; Iijima, H.; Saitow, A.; Fukumura, T.; Aoyama, Y.; Motoki, S.; Oikawa, T.; Yamazaki, T.; et al. Understanding hydrogen-bonding structures of molecular crystals via electron and NMR nanocrystallography. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Awosanya, E.O.; Lapin, J.; Nevzorov, A.A. NMR “crystallography” for uniformly (13C, 15N)-labeled oriented membrane proteins. Angew. Chem. Int. Ed. 2020, 59, 3554–3557. [Google Scholar] [CrossRef]
- Czernek, J.; Urbanová, M.; Brus, J. NMR crystallography of the polymorphs of metergoline. Crystals 2018, 8, 378. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, U.; Witter, R. Investigation of backbone dynamics and local geometry of bio-molecules using calculated NMR chemical shifts and anisotropies. J. Biomol. NMR 2019, 73, 727–741. [Google Scholar] [CrossRef]
- Narwani, T.J.; Santuz, H.; Shinada, N.; Vattekatte, A.M.; Ghouzam, Y.; Srinivasan, N.; Gelly, J.-C.; de Brevern, A.G. Recent advances on polyproline II. Amino Acids 2017, 49, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Czernek, J.; Pawlak, T.; Potrzebowski, M.J.; Brus, J. The comparison of approaches to the solid-state NMR-based structural refinement of vitamin B1 hydrochloride and of its monohydrate. Chem. Phys. Lett. 2013, 555, 135–140. [Google Scholar] [CrossRef]
- Perrin, B.S.; Pastor, R.W.; Cotton, M. Combining NMR Spectroscopic Measurements and Molecular Dynamics Simulations to Determine the Orientation of Amphipathic Peptides in Lipid Bilayers. In Advances in Biological Solid-State NMR: Proteins and Membrane-Active Peptides, 1st ed.; Separovic, S., Naito, A., Eds.; Royal Society of Chemistry: London, UK, 2014. [Google Scholar]
- Witanowski, M.; Stefaniak, L.; Szymański, S.; Januszewski, H. External neat nitromethane scale for nitrogen chemical shifts. J. Magn. Reson. 1977, 28, 217–226. [Google Scholar] [CrossRef]
- Alderman, D.W.; Sherwood, M.H.; Grant, D.M. Comparing, modeling and assigning chemical-shift tensors in the cartesian, irreducible spherical, and icosahedral representations. J. Magn. Reson. A 1993, 101, 188–197. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Theoretical predictions of the two-dimensional solid-state NMR spectra: A case study of the 13C—1H correlations in metergoline. Chem. Phys. Lett. 2013, 586, 56–60. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. The covariance of the differences between experimental and theoretical chemical shifts as an aid for assigning two-dimensional heteronuclear correlation solid-state NMR spectra. Chem. Phys. Lett. 2014, 608, 334–339. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Exploring accuracy limits of predictions of the 1-H chemical shielding anisotropy in the solid state. Molecules 2019, 24, 1731. [Google Scholar] [CrossRef] [Green Version]
- Ramon-Martín, F.; Annaval, T.; Buchoux, S.; Sarazin, C.; D’Amelio, N. ADAPTABLE: A comprehensive web platform of antimicrobial peptides tailored to the user’s research. Life Sci. Alliance 2020, 2, e201900512. [Google Scholar] [CrossRef] [Green Version]
- Chu, K.T.; Wang, H.X.; Ng, T.B. Fungal Peptides with Antifungal Activity. In Handbook of Biologically Active Peptides, 1st ed.; Kastin, A.J., Ed.; Academic Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Wel van der, P.C.A. New applications of solid-state NMR in structural biology. Emerg. Top. Life Sci. 2018, 2, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Marassi, F.M.; Opella, S.J. A Solid-State NMR Index of Helical Membrane Protein Structure and Topology. J. Magn. Reson. 2000, 144, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Salnikov, E.S.; Aisenbrey, C.; Pokrandt, B.; Brügger, B.; Bechinger, B. Structure, Topology, and Dynamics of Membrane-Inserted Polypeptides and Lipids by Solid-State NMR Spectroscopy: Investigations of the Transmembrane Domains of the DQ Beta-1 Subunit of the MHC II Receptor and of the COP I Protein. Front. Mol. Biosci. 2019, 6, 83. [Google Scholar] [CrossRef]
- Salnikov, E.S.; Friedrich, H.; Li, X.; Bertani, P.; Reissmann, S.; Hertweck, C.; O’Neil, J.D.J.; Raap, J.; Bechinger, B. Structure and Alignment of the Membrane-Associated Peptaibols Ampullosporin A and Alamethicin by Oriented 15N and 31P Solid-State NMR Spectroscopy. Biophys. J. 2009, 96, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Yarava, J.R.; Nishiyama, Y.; Raghothama, S.; Ramanathan, V.K. Conformational investigation of peptides using solid-state NMR spectroscopy–A study of polymorphism of β-turn peptides containing diprolines. Chem. Biol. Drug Des. 2020, 95, 394–407. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First principles simulation: Ideas, illustrations, and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculations of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. On the predictions of the B-11 solid state NMR parameters. Chem. Phys. Lett. 2016, 655, 66–70. [Google Scholar] [CrossRef]
Figure 1.
The Ramachandran plot for central glycyl residues in the set of tripeptides. Typical β-sheet, polyproline II (PP II) and αR-helical regions are schematically shown by large cyan, blue and green circles centered at [−150°; +150°], [−75°; +145°] and [90°; −30°], respectively.
Figure 1.
The Ramachandran plot for central glycyl residues in the set of tripeptides. Typical β-sheet, polyproline II (PP II) and αR-helical regions are schematically shown by large cyan, blue and green circles centered at [−150°; +150°], [−75°; +145°] and [90°; −30°], respectively.

Figure 2.
The isotropic 13C data of Cα nuclei of selected glycyls in tripeptides.

Figure 3.
The anisotropic 13C data of Cα nuclei of selected glycyls in tripeptides.

Figure 4.
The anisotropic 15N data of Namid nuclei of selected glycyls in tripeptides.

Figure 5.
The linear relationship between the 15Namid δii and σii data described in the text.

Figure 6.
The fragment of samarosporin I with visualized eigenvectors associated with the eigenvalues of the 15Namid chemical shielding tensor of Hyp13 residue (an eigenvector pertaining to the smallest, mid- and highest eigenvalue is colored in yellow, pink and dark green, respectively, and accordingly denoted as s1, s2 and s3).
Figure 6.
The fragment of samarosporin I with visualized eigenvectors associated with the eigenvalues of the 15Namid chemical shielding tensor of Hyp13 residue (an eigenvector pertaining to the smallest, mid- and highest eigenvalue is colored in yellow, pink and dark green, respectively, and accordingly denoted as s1, s2 and s3).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Peptides investigated in this work.
| Compound | Initial Geometry | SSNMR Data |
|---|---|---|
| Gly-Gly-Gly*HCl ‘GGG’ | 1110119 1 | from refs [12,13] |
| Val-Gly-Gly ‘VGG’ | 1129567 1 | from refs [12,13] |
| Pro-Gly-Gly ‘PGG’ | 1151185 1 | from refs [12,13] |
| Ala-Gly-Gly*H2O ‘AGG’ | 1119938 1 | from refs [12,13] |
| Phe-Gly-Gly ‘FGG’ | 1157783 1 | from refs [12,13] |
| Gly-Gly-Val*2H2O ‘GGV’ | 1134084 1 | from refs [12,13] |
| N-Ac-Aib-OH | 1130667 1 | from ref. [7] |
| N-Ac-Leu-OH | 624793 1 | from ref. [7] |
| Ala-Pro-Gly*H2O | 1160528 1 | from ref. [14] |
| melanostatin (see text) | 216376 1 | from ref. [17] |
| samarosporin I (see text) | 4G14 2 | predicted |
1 The Cambridge Crystallographic Database identifier (https://www.ccdc.cam.ac.uk/). 2 The Protein Data Bank identifier (https://www.rcsb.org/).
Table 2.
The chemical shift/shielding tensor data (in ppm) of the 15Namid nuclei in melanostatin. The σ values are taken from the CASTEP output, ε values are obtained from the parametrization as described in the text, and δ values are from the experiment [17].
Table 2.
The chemical shift/shielding tensor data (in ppm) of the 15Namid nuclei in melanostatin. The σ values are taken from the CASTEP output, ε values are obtained from the parametrization as described in the text, and δ values are from the experiment [17].
| Tensor Component | Site | |||
|---|---|---|---|---|
| Pro | Leu | Gly | ||
| the most shielded | σ | −11.8552 | −23.9238 | 134.6846 |
| ε | 220.6 | 231.9 | 83.5 | |
| δ | 215.1 | 229.9 | 88.2 | |
| the mid-shielded | σ | 152.3491 | 136.9511 | 172.5634 |
| ε | 67.0 | 81.4 | 48.1 | |
| δ | 74.1 | 88.1 | 45.9 | |
| the least shielded | σ | 163.1567 | 151.2735 | 218.4587 |
| ε | 56.9 | 68.0 | 5.1 | |
| δ | 54.4 | 61.2 | 4.1 | |
| isotropic part | σiso | 101.2169 | 88.1003 | 175.2326 |
| εiso | 114.8 | 127.1 | 45.6 | |
| δiso | 114.5 | 126.4 | 46.1 | |
Table 3.
The comparison of calculated and single-crystal (SC) solid-state NMR (SSNMR) (taken from reference [14] and shown in parentheses) angles describing an orientation of the 15Namid chemical shielding/shift tensor of the central residue (marked with an asterisk) in the peptide’s crystal frame. Designation of angles is the same as in reference [14] and should not be confused with the angles used in this work.
Table 3.
The comparison of calculated and single-crystal (SC) solid-state NMR (SSNMR) (taken from reference [14] and shown in parentheses) angles describing an orientation of the 15Namid chemical shielding/shift tensor of the central residue (marked with an asterisk) in the peptide’s crystal frame. Designation of angles is the same as in reference [14] and should not be confused with the angles used in this work.
| Site | Angle (in Degrees) 1 | ||
|---|---|---|---|
| γ | β | φ | |
| A G* G | 1.5 (1) | 18.8 (23) | 34.8 (36) |
| G G* V | 7.4 (11) | 23.4 (20) | 15.8 (15) |
| A P* G | 4.0 (5) | 23.3 (23) | 4.2 (5) |
1 ±3˚measurement uncertainty was reported in reference [14] for all the angles.
Table 4.
Selected parameters of the samarosporin I backbone (values in parentheses are from the XRD study [16]). The Namid–Hamid distances (rNH) are in picometers, all angles in degrees, and {ε11, ε22, ε33, εiso} data in ppm.
Table 4.
Selected parameters of the samarosporin I backbone (values in parentheses are from the XRD study [16]). The Namid–Hamid distances (rNH) are in picometers, all angles in degrees, and {ε11, ε22, ε33, εiso} data in ppm.
| Site | rNH | φ | ψ | εiso | ε11 | ε22 | ε33 | α | β | γ |
|---|---|---|---|---|---|---|---|---|---|---|
| Phe1 | 101.73 | −128 (−126) | −12 (−6) | 115.5 | 44.3 | 79.8 | 222.3 | 0.4 | 17.8 | 5.9 |
| Aib2 | 102.83 | −51 (−52) | −43 (−47) | 135.7 | 74.4 | 81.5 | 251.2 | 1.2 | 13.4 | 49.4 |
| Aib3 | 103.97 | −53 (−55) | −39 (−38) | 125.8 | 72.7ε | 76.2 | 228.6 | 1.3 | 18.0 | 35.2 |
| Aib4 | 102.50 | −55 (−55) | −47 (−50) | 121.5 | 58.9 | 81.0 | 224.5 | 4.2 | 16.3 | 33.7 |
| Val5 | 102.35 | −76 (−78) | −45 (−41) | 110.2 | 49.4 | 71.6 | 209.5 | 4.2 | 20.4 | 28.8 |
| Gly6 | 102.64 | −61 (−62) | −35 (−39) | 108.8 | 45.0 | 61.1 | 220.4 | 1.0 | 20.1 | 48.4 |
| Leu7 | 102.98 | −75 (−73) | −38 (−36) | 114.0 | 46.7 | 67.7 | 227.7 | 3.6 | 18.0 | 15.7 |
| Aib8 | 102.08 | −68 (−66) | −38 (−39) | 122.6 | 56.7 | 79.2 | 231.9 | 1.0 | 11.7 | 29.6 |
| Aib9 | 102.76 | −52 (−54) | −47 (−44) | 124.7 | 69.7 | 83.3 | 221.0 | 1.6 | 19.5 | 39.7 |
| Hyp10 | – | −65 (−64) | −13 (−16) | 127.6 | 43.7 | 119.8 | 219.2 | 1 | 1 | 1 |
| Gln11 | 103.01 | −87 (−87) | −11 (−11) | 107.2 | 48.7 | 59.3 | 213.6 | 2.6 | 20.6 | 44.5 |
| Iva12 | 102.72 | −54 (−53) | −41 (−40) | 123.8 | 63.5 | 77.6 | 230.4 | 1.1 | 17.2 | 19.4 |
| Hyp13 | – | −67 (−67) | −10 (−11) | 127.5 | 50.2 | 113.4 | 218.8 | 1 | 1 | 1 |
| Aib14 | 102.94 | −50 (−53) | −28 (−25) | 128.1 | 66.7 | 80.2 | 237.3 | 1.1 | 14.3 | 49.7 |
| Fol15 | 102.87 | −66 (−65) | – | 118.3 | 50.3 | 80.9 | 223.6 | 1.8 | 20.9 | 44.7 |
1 The definition of {α, β, γ} angles does not apply to hydroxyproline residues.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Czernek, J.; Brus, J. Monitoring the Site-Specific Solid-State NMR Data in Oligopeptides. Int. J. Mol. Sci. 2020, 21, 2700. https://doi.org/10.3390/ijms21082700
AMA Style
Czernek J, Brus J. Monitoring the Site-Specific Solid-State NMR Data in Oligopeptides. International Journal of Molecular Sciences. 2020; 21(8):2700. https://doi.org/10.3390/ijms21082700
Chicago/Turabian StyleCzernek, Jiří, and Jiří Brus. 2020. "Monitoring the Site-Specific Solid-State NMR Data in Oligopeptides" International Journal of Molecular Sciences 21, no. 8: 2700. https://doi.org/10.3390/ijms21082700
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.