Oxidative DNA Damage, Inflammatory Signature, and Altered Erythrocytes Properties in Diamond-Blackfan Anemia

 , and
, and
Abstract
:1. Introduction
2. Results
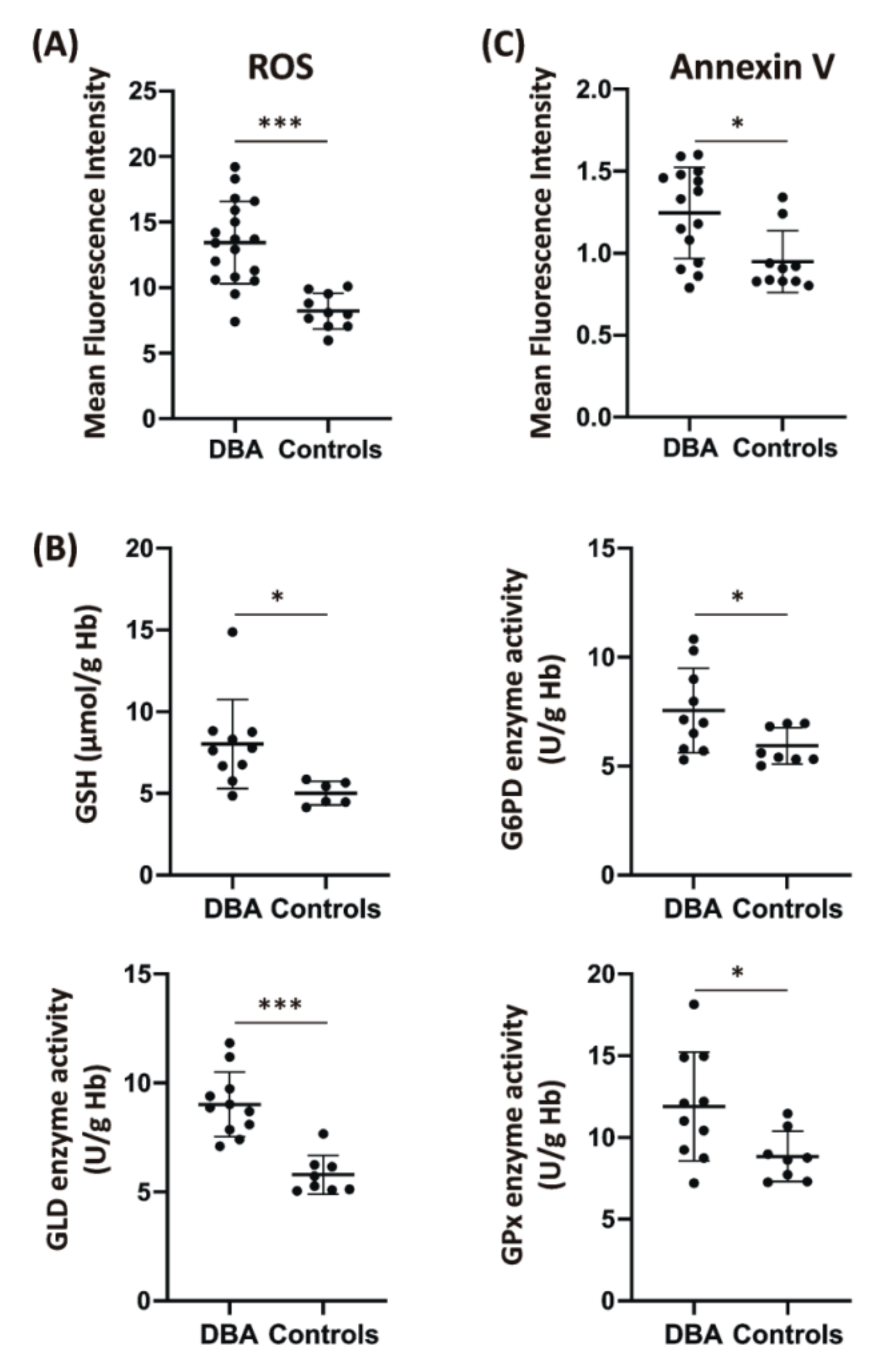
2.1. Characterization of Patients’ Cohort and Assessment of RBCs’ Oxidative Stress
2.2. Creation and Validation of DBA Cellular Model
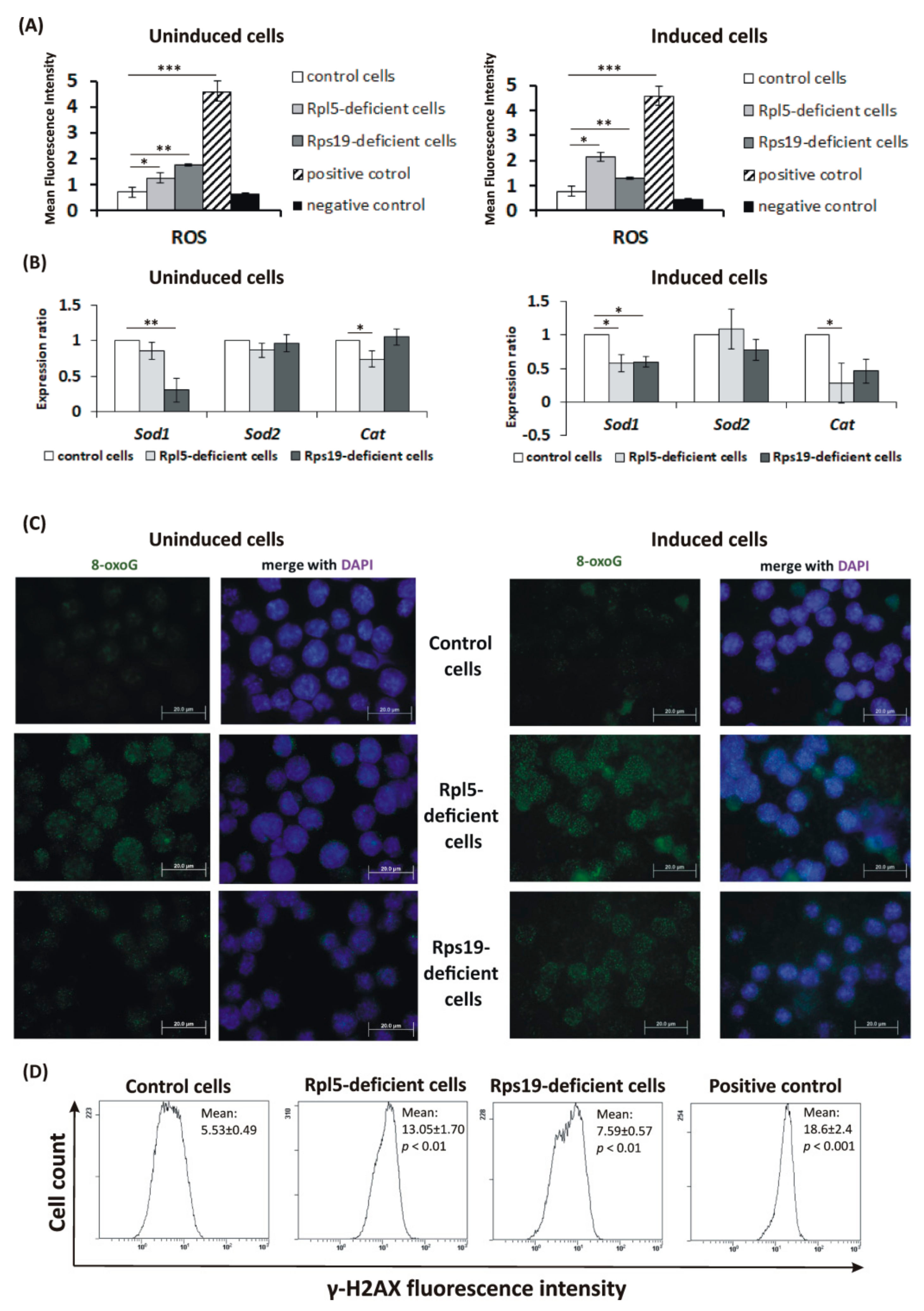
2.3. Oxidative DNA Damage in DBA Cellular Model
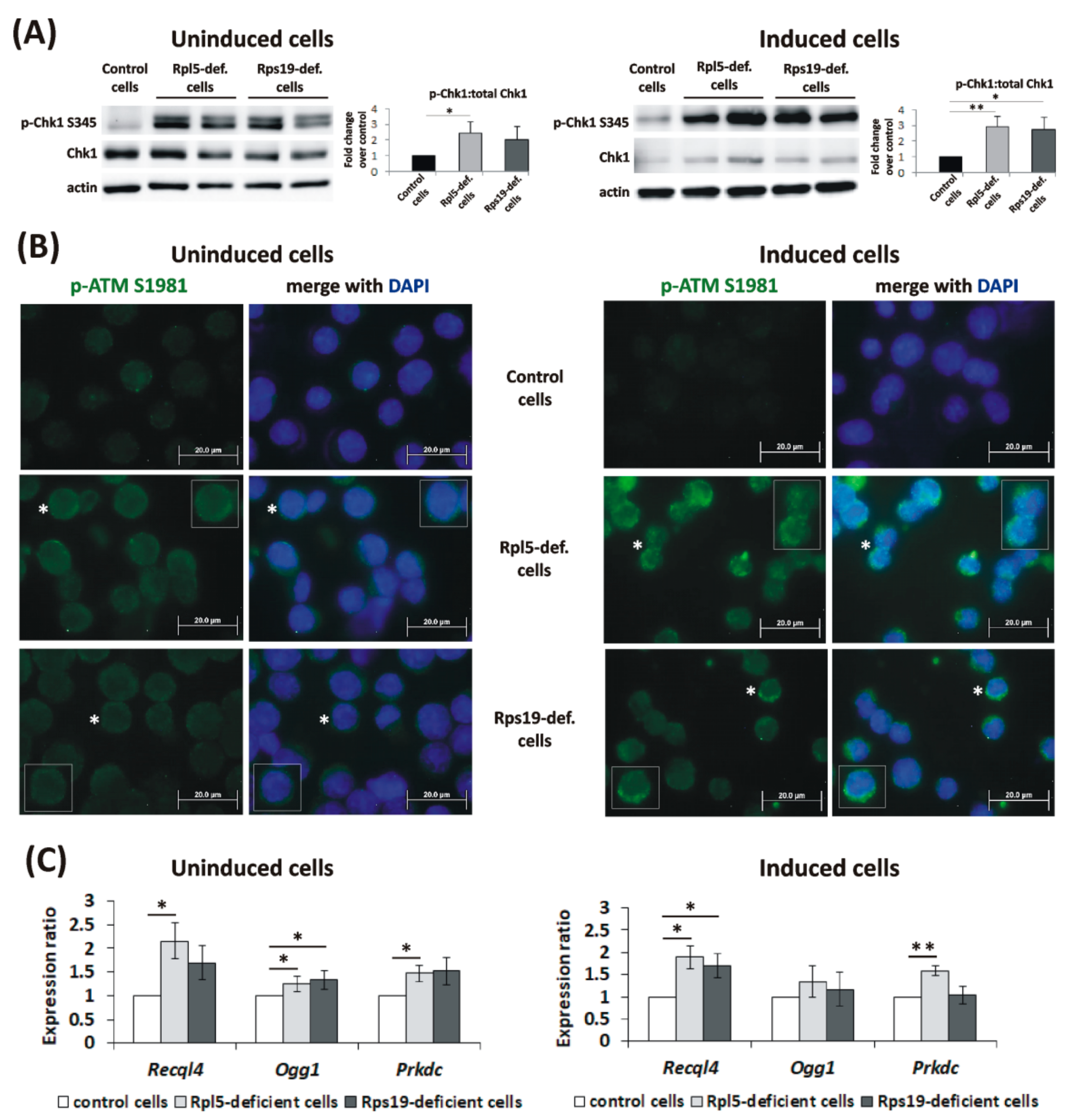
2.4. Activation of DDR Signaling and DNA Repair in DBA Cellular Model
2.5. Elevated Inflammatory Cytokines and Senescence in DBA Cellular Model
2.6. Oxidative DNA Damage, Elevated Inflammatory Cytokines, and Activated DDR Signaling in DBA Patients
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.1.1. Determination of ROS
4.1.2. Erythrocyte Annexin V Binding
4.1.3. Glutathione Measurements and Erythrocyte Enzyme Activity
4.1.4. Erythroblast Cell Culture and ICC Analyses
4.1.5. Determination of Inflammatory Cytokines Levels
4.1.6. Immunohistochemistry on Bone Marrow Samples
4.2. Cell Lines
4.2.1. Proliferation Assay and Apoptosis
4.2.2. Immunoblotting
4.2.3. Real-Time PCR Assay
4.2.4. Flow Cytometry Analysis of γ-H2AX and Phosphorylated p38
4.2.5. ROS Measurement
4.2.6. ICC of 8-OxoG and ATM pS1981
4.2.7. Cellular Senescence Activity Assay
4.2.8. Cell Cycle Analysis
4.3. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Da Costa, L.; Narla, A.; Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijnen, H.F.; van Wijk, R.; Pereboom, T.C.; Goos, Y.J.; Seinen, C.W.; van Oirschot, B.A.; van Dooren, R.; Gastou, M.; Giles, R.H.; van Solinge, W.; et al. Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway. PLoS Genet. 2014, 10, e1004371. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Bibikova, E.; Covey, T.M.; Nathanson, D.; Dimitrova, E.; Konto, Y.; Lindgren, A.; Glader, B.; Radu, C.G.; Sakamoto, K.M.; et al. The role of the DNA damage response in zebrafish and cellular models of Diamond Blackfan anemia. Dis. Model. Mech. 2014, 7, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Zidova, Z.; Kapralova, K.; Koralkova, P.; Mojzikova, R.; Dolezal, D.; Divoky, V.; Horvathova, M. DMT1-mutant Erythrocytes Have Shortened Life Span, Accelerated Glycolysis and Increased Oxidative Stress. Cell Physiol. Biochem. 2014, 34, 2221–2231. [Google Scholar] [CrossRef]
- Lang, F.; Abed, M.; Lang, E.; Föller, M. Oxidative Stress and Suicidal Erythrocyte Death. Antioxid. Redox Signal. 2014, 21, 138–153. [Google Scholar] [CrossRef]
- Utsugisawa, T.; Uchiyama, T.; Toki, T.; Ogura, H.; Aoki, T.; Hamaguchi, I.; Ishiguro, A.; Ohara, A.; Kojima, S.; Ohga, S.; et al. Erythrocyte glutathione is a novel biomarker of Diamond-Blackfan anemia. Blood Cells Mol. Dis. 2016, 59, 31–36. [Google Scholar] [CrossRef]
- Volejnikova, J.; Vojta, P.; Urbankova, H.; Mojzíkova, R.; Horvathova, M.; Hochova, I.; Cermak, J.; Blatny, J.; Sukova, M.; Bubanska, E.; et al. Czech and Slovak Diamond-Blackfan Anemia (DBA) Registry Update: Clinical Data and Novel Causative Genetic Lesions. Blood Cells Mol. Dis. 2020, 81, 102380. [Google Scholar] [CrossRef]
- Kempe, D.S.; Lang, P.A.; Duranton, C.; Akel, A.; Lang, K.S.; Huber, S.M.; Wieder, T.; Lang, F. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006, 20, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, M. Induction of Erythroid-Specific Expression in Murine Erythroleukemia (MEL) Cell Lines. Methods Mol. Biol. 1991, 7, 421–434. [Google Scholar] [PubMed]
- Ohene-Abuakwa, Y.; Orfali, K.A.; Marius, C.; Ball, S.E. Two-phase Culture in Diamond Blackfan Anemia: Localization of Erythroid Defect. Blood 2005, 105, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, J.M.; Kudisch, M.; Gross, R.; Nathan, D.G. Defective erythroid progenitor differentiation system in congenital hypoplastic (Diamond-Blackfan) anemia. Blood 1986, 67, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Pospisilova, D.; Holub, D.; Zidova, Z.; Sulovska, L.; Houda, J.; Mihal, V.; Hadacova, I.; Radova, L.; Dzubak, P.; Hajduch, M.; et al. Hepcidin levels in Diamond-Blackfan anemia reflect erythropoietic activity and transfusion dependency. Haematologica 2014, 99, e118–e121. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Zhao, B.; Tan, T.L.; Mei, Y.; Yang, J.; Yu, Y.; Verma, A.; Liang, Y.; Gao, J.; Ji, P. H2AX deficiency is associated with erythroid dysplasia and compromised haematopoietic stem cell function. Sci. Rep. 2016, 6, 19589. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Shamanna, R.A.; Keijzers, G.; Anand, R.; Rasmussen, L.J.; Cejka, P.; Croteau, D.L.; Bohr, V.A. RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell Rep. 2016, 16, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamanna, R.A.; Singh, D.K.; Lu, H.; Mirey, G.; Keijzers, G.; Salles, B.; Croteau, D.L.; Bohr, V.A. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 2014, 35, 2415–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.M.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef]
- Bibikova, E.; Youn, M.Y.; Danilova, N.; Ono-Uruga, Y.; Konto-Ghiorghi, Y.; Ochoa, R.; Narla, A.; Glader, B.; Lin, S.; Sakamoto, K.M. TNF-mediated inflammation represses GATA1 and activates p38 MAP kinase in RPS19-deficient hematopoietic progenitors. Blood 2014, 124, 3791–3798. [Google Scholar] [CrossRef]
- Schieven, G.L. The p38alpha kinase plays a central role in inflammation. Curr. Top. Med. Chem. 2009, 9, 1038–1048. [Google Scholar] [CrossRef]
- Xu, D.; Matsumoto, M.L.; McKenzie, B.S.; Zarrin, A.A. TPL2 kinase action and control of inflammation. Pharmacol. Res. 2018, 129, 188–193. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Corral, L.G.; Haslett, P.A.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential Cytokine Modulation and T Cell Activation by Two Distinct Classes of Thalidomide Analogues That Are Potent Inhibitors of TNF-alpha. J. Immunol. 1999, 163, 380–386. [Google Scholar] [PubMed]
- Escoubet-Lozach, L.; Lin, I.L.; Jensen-Pergakes, K.; Brady, H.A.; Gandhi, A.K.; Schafer, P.H.; Muller, G.W.; Worland, P.J.; Chan, K.W.H.; Verhelle, D. Pomalidomide and Lenalidomide Induce p21 WAF-1 Expression in Both Lymphoma and Multiple Myeloma Through a LSD1-mediated Epigenetic Mechanism. Cancer Res. 2009, 69, 7347–7356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horos, R.; Ijspeert, H.; Pospisilova, D.; Sendtner, R.; Andrieu-Soler, C.; Taskesen, E.; Nieradka, A.; Cmejla, R.; Sendtner, M.; Touw, I.P.; et al. Ribosomal Deficiencies in Diamond-Blackfan Anemia Impair Translation of Transcripts Essential for Differentiation of Murine and Human Erythroblasts. Blood 2012, 119, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Walker, C.L. Differential localization of ATM is correlated with activation of distinct downstream signaling pathways. Cell Cycle 2010, 9, 3685–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.C.; Said, A.; Corcuera, D.; McLaughlin, D.; Kell, P.; Doctor, A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. FASEB J. 2009, 23, 3159–3170. [Google Scholar] [CrossRef] [Green Version]
- Bester, J.; Pretorius, E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188. [Google Scholar] [CrossRef]
- Aspesi, A.; Pavesi, E.; Robotti, E. Dissecting the Transcriptional Phenotype of Ribosomal Protein Deficiency: Implications for Diamond-Blackfan Anemia. Gene 2014, 545, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal Protein L11 Mutation in Zebrafish Leads to Haematopoietic and Metabolic Defects. Br. J. Haematol. 2011, 152, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Alexander, A.; Cai, S.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. The Ribosomal RPL10 R98S Mutation Drives IRES-dependent BCL-2 Translation in T-ALL. Leukemia 2019, 33, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Sulima, S.O.; Kampen, K.R.; Vereecke, S.; Pepe, D.; Fancello, L.; Verbeeck, J.; Dinman, J.D.; De Keersmaecker, K. Ribosomal Lesions Promote Oncogenic Mutagenesis. Cancer Res. 2019, 79, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstrom, M.S.; Jin, A.; Deisenroth, C.; White Wolf, G.; Zhang, Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell Biol. 2007, 27, 1056–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestov, D.G.; Strezoska, Z.; Lau, L.F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef] [Green Version]
- Matsui, K.; Giri, N.; Alter, B.P.; Pinto, L.A. Cytokine production by bone marrow mononuclear cells in inherited bone marrow failure syndromes. Br. J. Haematol. 2013, 163, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Danilova, N.; Wilkes, M.; Bibikova, E.; Youn, M.Y.; Sakamoto, K.M.; Lin, S. Innate immune system activation in zebrafish and cellular models of Diamond Blackfan Anemia. Sci. Rep. 2018, 8, 5165. [Google Scholar] [CrossRef]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef]
- Gudkov, A.V.; Gurova, K.V.; Komarova, E.A. Inflammation and p53. A Tale of Two Stresses. Genes Cancer 2011, 2, 503–516. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ali, A.; Junaid, M.; Liu, C.; Kaushik, A.C.; Cho, W.C.S.; Wei, D.Q. Identification of novel drug targets for diamond-blackfan anemia based on RPS19 gene mutation using protein-protein interaction network. BMC Syst. Biol. 2018, 12, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jason, J.; Archibald, L.K.; Nwanyanwu, O.C.; Byrd, M.G.; Kazembe, P.N.; Dobbie, H.; Jarvis, W.R. Comparison of serum and cell-specific cytokines in humans. Clin. Diagn. Lab. Immunol. 2001, 8, 1097–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.A.; Waddelow, T.A.; Caro, J.; Oliff, A.; Roodman, G.D. Chronic exposure to tumor necrosis factor in vivo preferentially inhibits erythropoiesis in nude mice. Blood 1989, 74, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Means, R.T., Jr.; Dessypris, E.N.; Krantz, S.B. Inhibition of human erythroid colony-forming units by interleukin-1 is mediated by gamma interferon. J. Cell Physiol. 1992, 150, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.H.; Price, J.O.; Brunner, T.; Krantz, S.B. Fas ligand is present in human erythroid colony-forming cells and interacts with Fas induced by interferon γ to produce erythroid cell apoptosis. Blood 1998, 91, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Li, J.; Sejas, D.P.; Zhang, X.; Qiu, Y.; Nattamai, K.J.; Rani, R.; Rathbun, K.R.; Geiger, H.; Williams, D.A.; Bagby, G.C.; et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J. Clin. Investig. 2007, 117, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Amer, J.; Dana, M.; Fibach, E. The antioxidant effect of erythropoietin on thalassemic blood cells. Anemia 2010, 2010, 978710. [Google Scholar] [CrossRef] [Green Version]
- Beutler, E.; Blume, K.G.; Kaplan, J.C.; Löhr, G.W.; Ramot, B.; Valentine, W.N. International Committee for Standardization in Haematology: Recommended methods for red-cell enzyme analysis. Br. J. Haematol. 1977, 35, 331–340. [Google Scholar] [CrossRef]
- Mojzikova, R.; Dolezel, P.; Pavlicek, J.; Mlejnek, P.; Pospisilova, D.; Divoky, V. Partial glutathione reductase deficiency as a cause of diverse clinical manifestations in a family with unstable hemoglobin (Hemoglobin Haná, β63(E7) His-Asn). Blood Cells Mol. Dis. 2010, 45, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Mojzikova, R.; Koralkova, P.; Holub, D.; Zidova, Z.; Pospisilova, D.; Cermak, J.; Striezencova Laluhova, Z.; Indrak, K.; Sukova, M.; Partschova, M.; et al. Iron status in patients with pyruvate kinase deficiency: Neonatal hyperferritinaemia associated with a novel frameshift deletion in the PKLR gene (p.Arg518fs), and low hepcidin to ferritin ratios. Br. J. Haematol. 2014, 165, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, A.; Nussenzveig, R.; Liu, E.; Gottshalk, S.; Chang, K.; Prchal, J.T. In vitro expansion of erythroid progenitors from polycythemia vera patients leads to decrease in JAK2 V617F allele. Exp. Hematol. 2007, 35, 587–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leberbauer, C.; Boulme, F.; Unfried, G.; Huber, J.; Beug, H.; Mullner, E.W. Different steroids co-regulate long-term expansion versus terminal differentiation in primary human erythroid progenitors. Blood 2005, 105, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Stetka, J.; Vyhlidalova, P.; Lanikova, L.; Koralkova, P.; Gursky, J.; Hlusi, A.; Flodr, P.; Hubackova, S.; Bartek, J.; Hodny, Z.; et al. Addiction to DUSP1 protects JAK2V617F-driven polycythemia vera progenitors against inflammatory stress and DNA damage, allowing chronic proliferation. Oncogene 2019, 38, 5627–5642. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.W.; Chen, R.; Huang, S.Y.; Corral, L.G.; Wong, L.M.; Patterson, R.T.; Chen, Y.; Kaplan, G.; Stirling, D.I. Amino-substituted thalidomide analogs: Potent inhibitors of TNF-α production. Bioorg. Med. Chem. Lett. 1999, 9, 1625–1630. [Google Scholar] [CrossRef]
- Koledova, Z.; Kafkova, L.R.; Calabkova, L.; Krystof, V.; Dolezel, P.; Divoky, V. Cdk2 inhibition prolongs G1 phase progression in mouse embryonic stem cells. Stem Cells Dev. 2010, 19, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Horvathova, M.; Kapralova, K.; Zidova, Z.; Dolezal, D.; Pospisilova, D.; Divoky, V. Erythropoietin-driven signaling ameliorates the survival defect of DMT1-mutant erythroid progenitors and erythroblasts. Haematologica 2012, 97, 1480–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pct | Sex | Age | Gene Mutation | Treatment | ROS | Annexin V | ELISA Inflamm. Cytokines | GSH Measurements | RBC Enzyme Activities | In Vitro Cultivation | IHC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | F | 54 | RPL5 | R | + | + | + | + | NA | + | NA |
| P2 * | M | 32 | RPL5 | R | + | + | + | + | + | + | + |
| P3 | F | 42 | RPL5 | R | NA | + | + | + | + | NA | NA |
| P4 * | M | 33 | RPL5 | R | + | + | + | + | + | + | + |
| P5 | M | 20 | RPS19 | S | + | + | + | NA | NA | NA | NA |
| P6 | F | 19 | RPS19 | S | + | + | + | NA | NA | NA | NA |
| P7 | M | 40 | RPS19 | R | + | + | + | + | + | + | NA |
| P8 | M | 46 | RPS19 | S, Leu | + | NA | + | + | + | + | NA |
| P9 * | M | 28 | RPS19 | R | + | + | NA | + | + | NA | + |
| P10 | F | 46 | RPL11 | R | + | NA | + | NA | NA | NA | NA |
| P11 | M | 8 | RPL11 | S | + | + | NA | NA | NA | NA | NA |
| P12 | F | 36 | RPL11 | R | + | NA | NA | NA | NA | NA | NA |
| P13 | F | 54 | RPL11 | S | + | NA | NA | NA | NA | NA | NA |
| P14 | F | 27 | RPS7 | R | + | + | + | + | + | NA | NA |
| P15 | F | 41 | RPS7 | R | + | + | NA | + | + | NA | NA |
| P16 | F | 54 | RPS7 | R | + | + | NA | + | + | NA | NA |
| P17 | F | 33 | delRPL35a | S | + | + | NA | NA | NA | NA | NA |
| P18 | M | 34 | No mut | S | + | + | NA | + | + | NA | + |
| P19 | M | 10 | No mut | R | + | + | + | NA | NA | NA | NA |
| P20 | F | 30 | RPS19 | T, DRX | NA | NA | NA | NA | NA | NA | + |
| P21 | F | 34 | RPS19 | T, DRX | NA | NA | NA | NA | NA | NA | + |
| P22 | M | 26 | RPS26 | T, DRX | NA | NA | NA | NA | NA | NA | + |
| P23 | M | 21 | RPS26 | T, DRX | NA | NA | NA | NA | NA | NA | + |
| P24 § | F | 31§ | RPL11 | T, DRX | NA | NA | NA | NA | NA | NA | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapralova, K.; Jahoda, O.; Koralkova, P.; Gursky, J.; Lanikova, L.; Pospisilova, D.; Divoky, V.; Horvathova, M. Oxidative DNA Damage, Inflammatory Signature, and Altered Erythrocytes Properties in Diamond-Blackfan Anemia. Int. J. Mol. Sci. 2020, 21, 9652. https://doi.org/10.3390/ijms21249652
Kapralova K, Jahoda O, Koralkova P, Gursky J, Lanikova L, Pospisilova D, Divoky V, Horvathova M. Oxidative DNA Damage, Inflammatory Signature, and Altered Erythrocytes Properties in Diamond-Blackfan Anemia. International Journal of Molecular Sciences. 2020; 21(24):9652. https://doi.org/10.3390/ijms21249652
Chicago/Turabian StyleKapralova, Katarina, Ondrej Jahoda, Pavla Koralkova, Jan Gursky, Lucie Lanikova, Dagmar Pospisilova, Vladimir Divoky, and Monika Horvathova. 2020. "Oxidative DNA Damage, Inflammatory Signature, and Altered Erythrocytes Properties in Diamond-Blackfan Anemia" International Journal of Molecular Sciences 21, no. 24: 9652. https://doi.org/10.3390/ijms21249652