Parametrizing the Spatial Dependence of 1H NMR Chemical Shifts in π-Stacked Molecular Fragments


Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovsky Square #2, 16206 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(21), 7908; https://doi.org/10.3390/ijms21217908
Submission received: 14 October 2020
/
Revised: 20 October 2020
/
Accepted: 22 October 2020
/
Published: 24 October 2020
(This article belongs to the Special Issue Molecular Structure and Dynamics Probed by Spectroscopic Techniques and Computational Approaches: New Trends by NMR, FTIR, Neutron Scattering and Simulation)
Abstract
:Most recently a renewed interest in several areas has arisen in factors governing the 1H NMR chemical shift (1H CS) of protons in aromatic systems. Therefore, it is important to describe how 1H CS values are affected by π-stacking intermolecular interactions. The parametrization of radial and angular dependences of the 1H CS is proposed, which is based on conventional gauge-independent atomic orbital (GIAO) calculations of explicit molecular fragments. Such a parametrization is exemplified for a benzene dimer with intermonomer vertical and horizontal distances which are in the range of values often found in crystals of organic compounds. Results obtained by the GIAO calculations combined with B3LYP and MP2 methods were compared, and revealed qualitatively the same trends in the 1H CS data. The parametrization was found to be quantitatively correct for the T-shaped benzene dimers, and its limitations were discussed. Parametrized 1H CS surfaces should become useful for providing additional restraints in the search of site-specific information through an analysis of structurally induced 1H CS changes.
1. Introduction
The proton nuclear magnetic resonance (1H NMR) is a crucial technique for solving a vast variety of chemical, physical, and biological problems in solution [1] and solid phases [2]. Some of those problems concern the dispersive interactions [3] between aromatic moieties and the role they play in various phenomena (see references [4,5,6,7] for the most recent examples). In such studies, the key parameter is the 1H chemical shift (1H CS) perturbation caused by the presence of aromatic fragments in the vicinity of an investigated proton [8,9]. It should be noted that the 1H CS of protons in aromatic molecules has received considerable interest, as most recently summarized in references [10,11]. Therefore, it is desirable to study factors governing the 1H CS values in molecules which exhibit C–H/π aromatic stacking interactions. Here, conventional gauge-independent atomic orbital (GIAO) calculations (see Materials and Methods) are carried out for the proton sites in two types of stacked molecular dimers. The first type are benzene dimers which serve for the purpose of a parametrization of the 1H CS landscape, as detailed in Section 2.1, and for checking the precision of the proposed parametrization. The second type of investigated dimers are those taken from crystals of midsized organic molecules [12,13] (see Section 2.2). For these systems, accurate 1H CS data are available from the solid-state NMR (SSNMR) measurements [12,14], and hence they are used in reliability considerations. In particular, the GIAO 1H CS estimates for the dimers are confronted with experiment and with results of the gauge-including projector augmented wave (GIPAW) calculations performed in a plane-wave density functional theory (PW DFT) scheme for periodic structures of aforementioned crystals [15]. In this way, it has been shown that it should be possible to successfully apply the parametrization process to other ligands and to configurations relevant for binding of those ligands. At the same time, an inherent limitation of this approach should be noted, which is the presence of a generally shaped contour line for a given 1H CS value, as discussed in Section 3 and visualized. However, the parametrized 1H CS surfaces are expected to be applied together with other structural information provided by NMR and possibly diffraction measurements, or by advanced computational methods [16], which would constrain stacking fragments in the correct spatial arrangement compatible with some specific 1H CS value. Importantly, a set of 1H CS values could, then, be used to elucidate structures of complex systems, for instance, polydopamine [17,18,19,20]. It should also be noted that the 1H CS surfaces can be obtained with an inclusion of solvation at the interacting sites [21] in order to properly describe binding between organic ligands and models of large biologically active molecules in solution.
2. Results
2.1. Development of the Proton Chemical Shift (1H CS) Surface
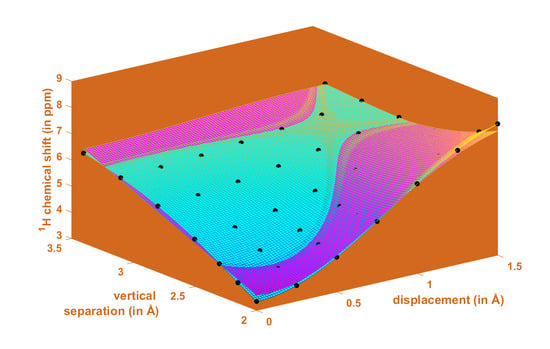
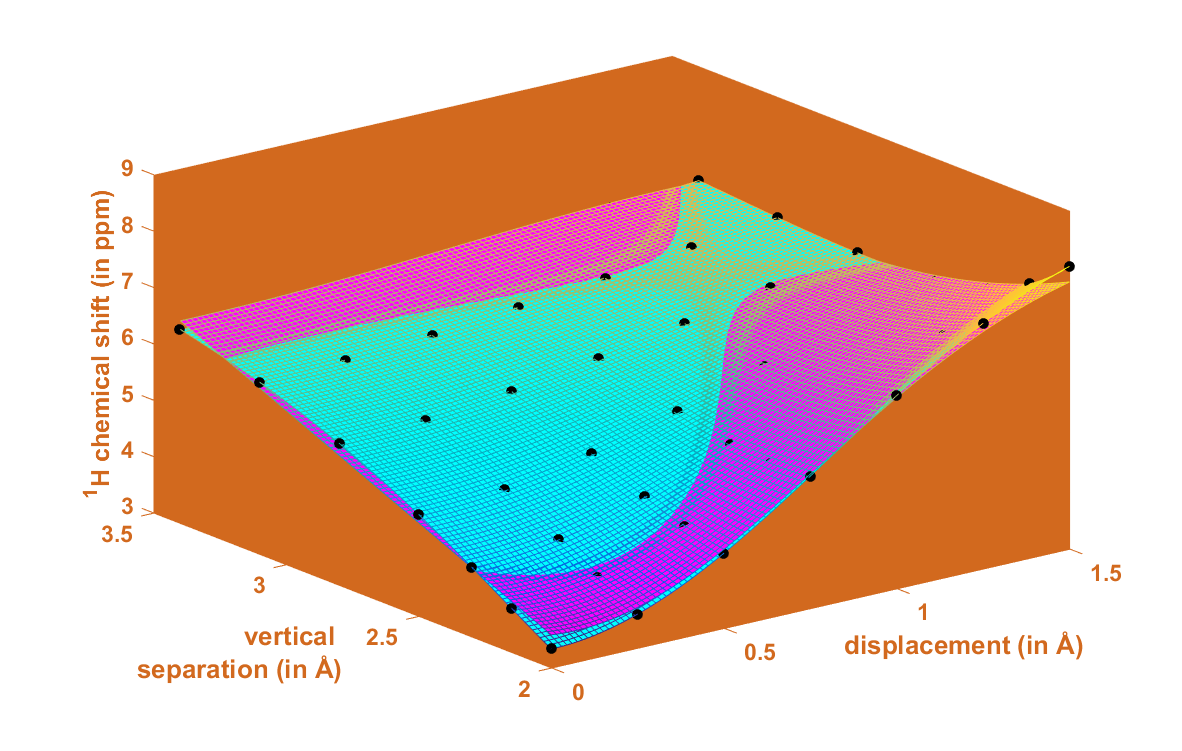
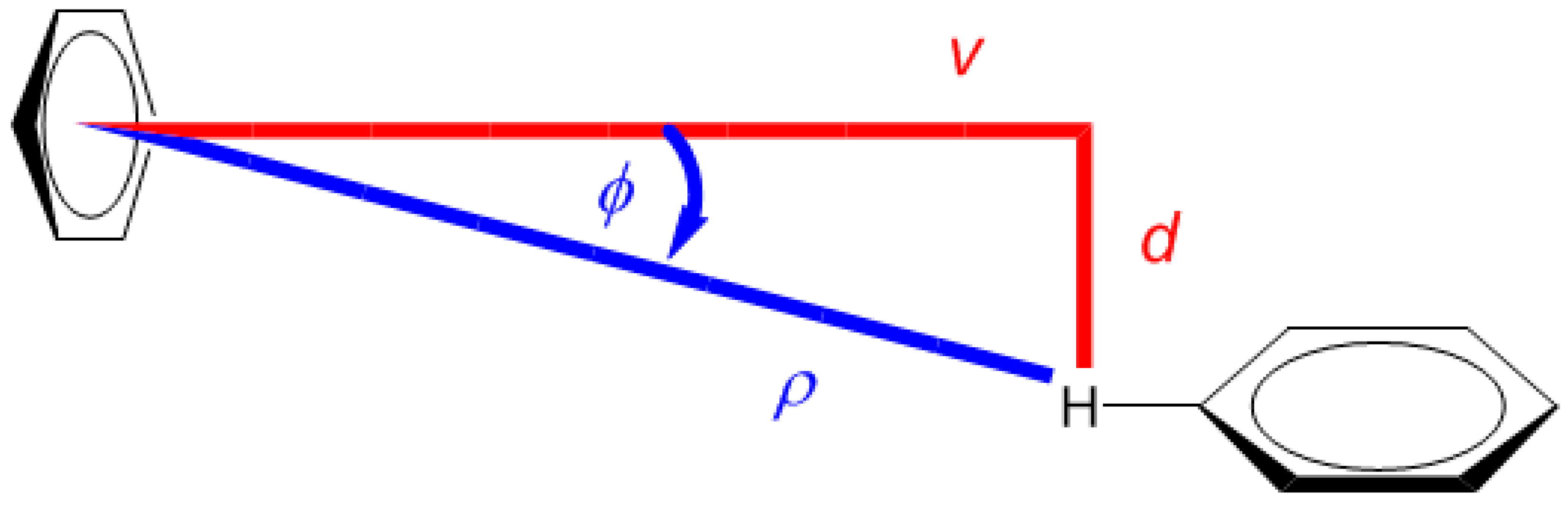
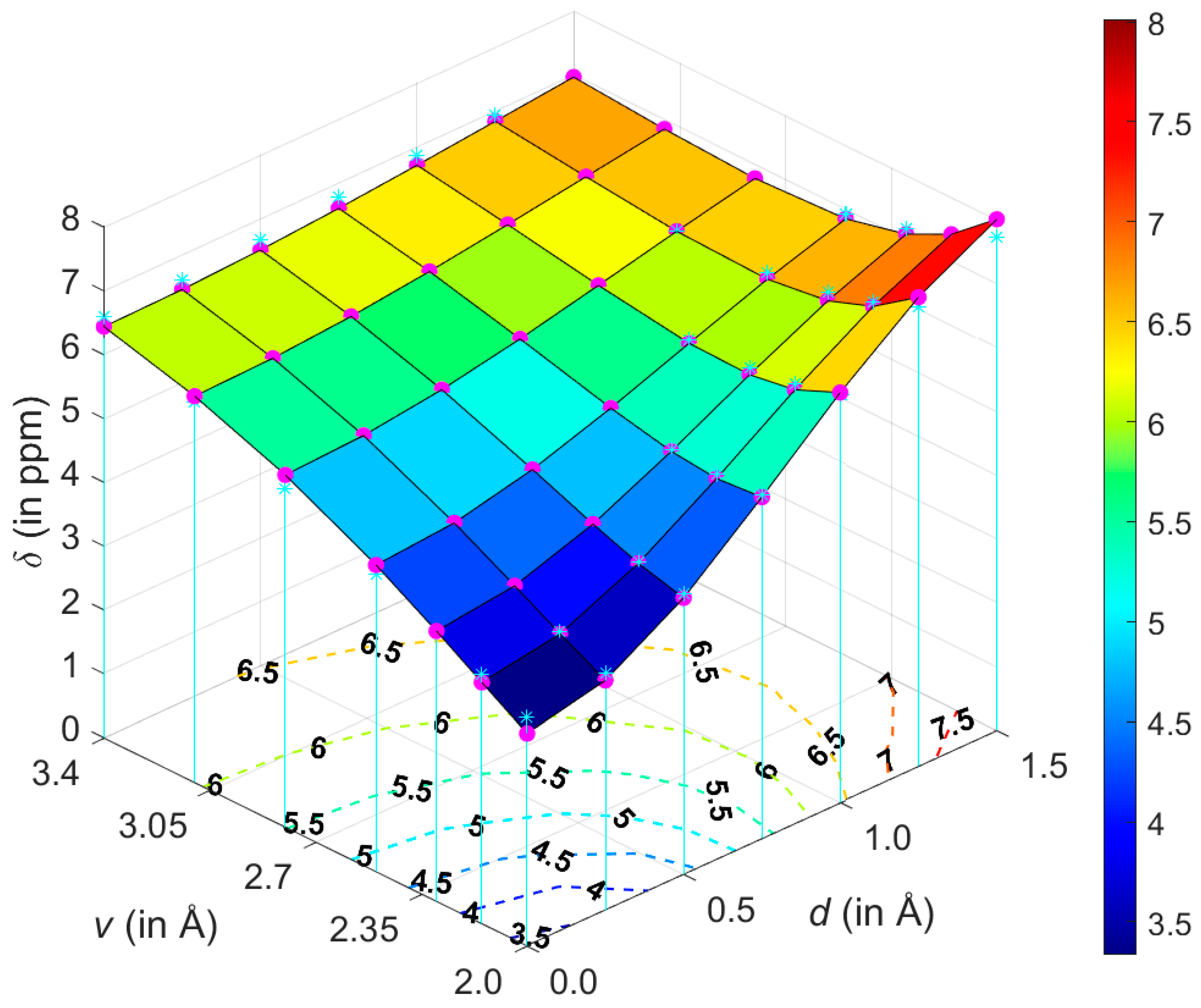
The energy parameters of clusters of benzene in various media have been intensely studied [22,23]. The potential energy surface (PES) of the gas-phase benzene dimer is quite well understood [24]. Here, for the purpose of describing the landscape of the 1H CS values, the T-shaped dimer in “C2v over atom” configuration was adopted from the work of Head-Gordon at al. [25] (the energetics are discussed in Section 2.3 and put into context of other configurations). Using this structure, the grid consisting of 7 × 7 points was constructed by simultaneously varying two parameters, without any relaxation of the geometry. These parameters were (i) v, the vertical separation between the investigated proton and the center of the other monomer and (ii) d, the lateral displacement in the plane parallel to the monomer not containing that proton (see Figure 1). It should be mentioned that one of the grid points corresponded to the geometry directly taken from reference [25], with v = 2.302 Å and d = 0.0 Å, and that all structures with nonzero values of d were of Cs symmetry. For the points from intervals of v between 2.002 and 3.402 Å and of d between 0.0 and 1.5 Å, the 1H CS isotropic chemical shielding value of the investigated proton was provided by the GIAO and GIAO and Becke’s three-parameter together with Lee–Yang–Parr functionals (GIAO-MP2)/6-311++G(2d,2p), and GIAO and Becke’s three-parameter, Lee–Yang–Parr (GIAO-B3LYP)/6-311++G(2d,2p) approaches (see Materials and Methods). These two methods were successfully applied to investigate trends in the chemical shielding [26,27,28]. Estimates of the 1H CS values, denoted as δ, were obtained using the 1H isotropic chemical shielding calculated by the corresponding method for a proton of tetramethylsilane molecule optimized at the MP2/aug-cc-pVTZ level, as detailed in Section 4.
An attempt was made to analytically describe the dependence of δ upon the variables v and d. At first, however, geometries were expressed in the polar coordinates (ρ, ϕ), also depicted in Figure 1. Numerical experiments were, then, performed in this coordinate system using relevant toolboxes of Matlab®, and it was found that both sets of input δ data could be successfully fitted to a relatively simple functional form given by Equation (1). Subsequently, the fitting was repeated in the (v, d) coordinate system while employing “e04fcf” subroutine from NAG® Library (related f90 program also used “lsqgrd” subroutine to check that the gradients of , taken with respect to {A, B, C, D, E, F} parameters of Equation (1), were sufficiently close to zero). The resulting parameter values were considered to be final. They are listed in Supplementary Materials Table S1, and both sets of 49 δ points are available from Table S4. The sole idea behind finding this analytic expression for δ is to obtain a formula which is sufficiently accurate for numerically describing the 1H CS surfaces, and Equation (1) should not be interpreted in any other way. It follows from a comparison of the GIAO-MP2 and GIAO-B3LYP surfaces that these methods provided qualitatively the same 1H CS landscape. This is significant, since the GIAO-B3LYP/6-311++G(2d,2p) approach, contrary to its MP2 counterpart, can be routinely applied to large molecular fragments, possibly containing more than one hundred atoms. Satisfactory precision of the fitting is also important, i.e., the maximum absolute deviation, average absolute deviation, residual norm, and x2 are 0.28 ppm, 0.10 ppm, 0.83 ppm, and 0.1247, respectively, for the GIAO-MP2 data, while these values accordingly amount to 0.32, 0.09, 0.61 ppm and 0.0993 for the GIAO-B3LYP data.
Figure 2 graphically presents an example 1H CS surface and its fairly complex curvature and other issues are discussed in Section 3, while reliability of the model is addressed below.
This description of a spatial arrangement of benzene monomers using only two geometry variables is, of course, simplified. Hence, it is of importance to assess how accurate the 1H CS parametrization would be if applied to the benzene dimer configurations with mutual orientation of planes other than the one shown in Figure 1. This assessment was performed for the tilted T-shaped structures from [29], which are available from the BEGDB database [30] (their dissociation curve is analyzed in Section 2.3). The v, d values of five of those geometries fall into the parametrized interval, and therefore their coordinates were used to obtain the 1H CS from the model and directly from the GIAO calculations. The predicted 1H chemical shielding data are summarized in Table S2 and show only small differences between the parametrized values and their counterparts provided explicitly by quantum chemical calculations (maximum discrepancy amounts to 0.28 ppm and is found for the MP2 data of the structure with v = 2.6595 Å and d = 0.1361 Å). Moreover, these datasets exhibit the same geometry dependence, namely, the 1H chemical shielding values decrease with an increase in v and d in this investigated region. The above two findings show that the presented model also works well for the benzene dimers featuring interplanar angles which have not been included in the parameterization. Therefore, it seems that it might be possible to use only two spatial coordinates to reliably quantify how π-stacking interactions affect the 1H CS of protons in aromatic systems in general. However, it should be kept in mind that this model relies on the ability of quantum chemical calculations to accurately describe the pair interactions influencing the 1H CS value in a molecular cluster. Hence, in Section 2.2, this ability is evaluated for organic solids whose structures pose additional challenges due to crystal packing and due to heteroatoms or substituents present in the aromatic ring that also contains the investigated proton.
2.2. Validation of the Dimer Model
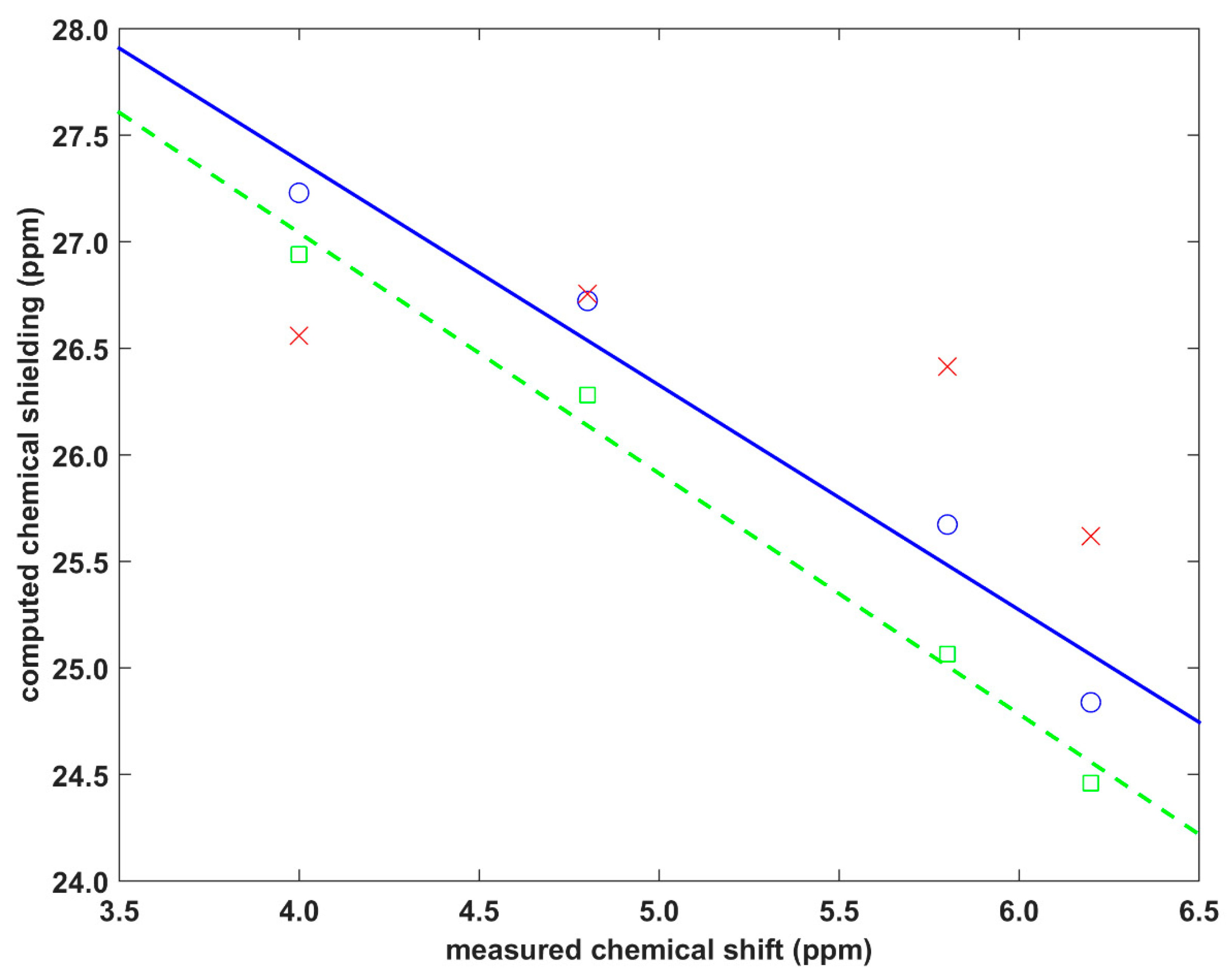
Older research on the 1H CS of stacked proton sites in molecular crystals has been presented in the excellent review article [31]. Here, two solid phase systems are analyzed, which have been carefully studied by the groups of Kentgens [14] and Brown [12], and thus they provide the benchmark data. Their data are considered to be fully reliable and cover a relatively large interval of pertinent 1H CS values (see Table 1). The first system considers the protons, numbered H10′ and H11′ and shown in Figure 11b of [14], of the crystalline isocyanoalanyl carbazole amid [13]. The second system concerns the 1:1 cocrystal of dithianon and pyrimethanil with protons numbered H2 and H25 and shown in Figure 5 of [12]. Experimental and theoretical data are collected in Table 1 (the following values of the 1H absolute isotropic chemical shielding of a proton in tetramethylsilane were used for referencing: 30.8868 ppm for GIPAW and Perdew-Burke-Erzerhof (GIPAW-PBE) and 31.8308 ppm for GIAO-B3LYP, respectively). The investigated protons are practically unaffected by the C–H…X hydrogen bonding, which is not the case, for example, for aromatic protons in the well-known 1:1 cocrystal of indomethacin and nicotinamide [32,33], or in some systems previously studied by this authors, in particular, L-tyrosine hydrochloride [34]. Consequently, the investigated protons are supposed to probe an influence of the C–H/π stacking only and are used for further testing of the present approach (see Figure 3; computational estimates of the 1H CS are plotted against experimental values in Figure S2). For this type of environment, it was ascertained that the GIAO-B3LYP/6-311++G(2d,2p) dimer calculations could provide accurate 1H chemical shielding values, i.e., the standard deviation of residuals and adjusted R2 of the corresponding linear regression are 0.21 ppm and 0.93, respectively. Accordingly, these results amount to 0.12 ppm and 0.98 for the periodic GIPAW calculations which are, thus, highly successful, as was anticipated on the basis of previous work on structurally similar systems, for instance, naproxen [35,36]. It has to be mentioned, however, that large errors of the PW DFT predicted 1H CS may occur for hydroxyl hydrogen-bonded sites [37,38,39], but those discrepancies are caused mainly by the proton dynamics [40,41,42] that is unlikely to be significant in the molecular crystals studied here. Nevertheless, the current results validate the approach which applies the GIAO-B3LYP calculations of dimers for describing the spatial dependence of the 1H CS in fragments affected by π-stacking in molecular solids.
Corresponding results obtained from the model expressed by Equation (1) are also plotted in Figure 3. Their quality is generally poor, as the parametrization was carried out using the benzene dimer, not for fragments actually present in the investigated crystals. However, an inspection of these results indicates that the parameters of Equation (1) may be transferable to structurally similar residues. This can be seen for the proton site involved in the C–H/π interaction between fused (and otherwise unsubstituted) benzene rings. Namely, the H11′ site of the isocyanoalanyl carbazole amid structure [13] is located on one such ring (denoted as “P” in [14]) of a carbazole unit, and is stacked by another benzene ring (also “P”) of the neighboring molecule. As shown in Figure 3, at the 1H CS of 4.8 ppm, the difference between the parametrized result and the GIAO-B3LYP value is minimal (0.04 ppm). The contacts of the remaining protons (H11′, H2, and H25 of the structures specified above) involve substituted rings and are not detailed here. Separate parametrizations would be needed in order to accurately describe the 1H CS in these moieties. It should be kept in mind that the presented analysis is implicitly influenced by the 1H CS measurement uncertainties. Those uncertainties can be expected to be about 0.2 ppm, but they could be reduced, in particular, by using ultrafast magic-angle spinning SSNMR techniques [43,44,45], and at very high magnetic fields.
2.3. Dimerization Energy Considerations
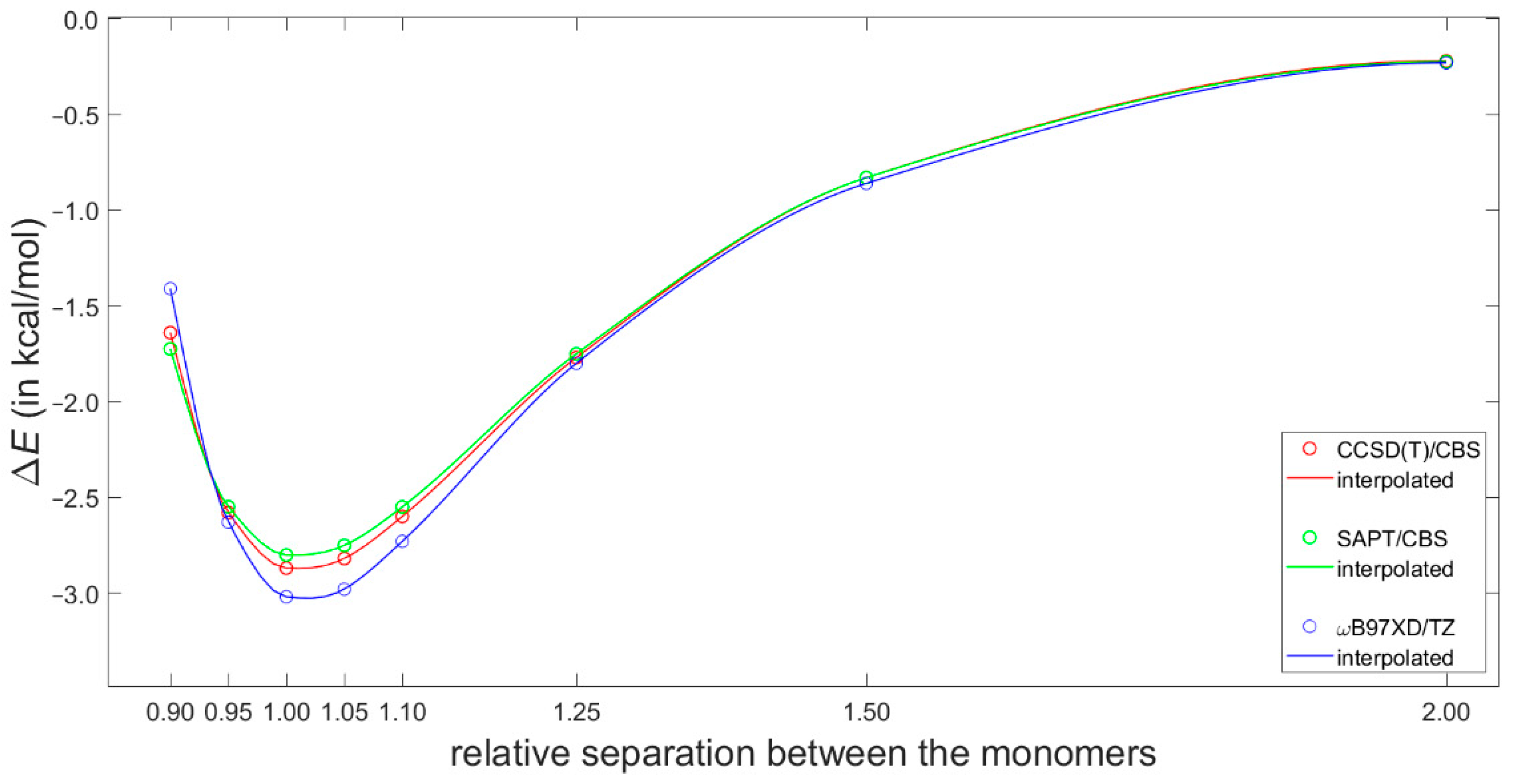
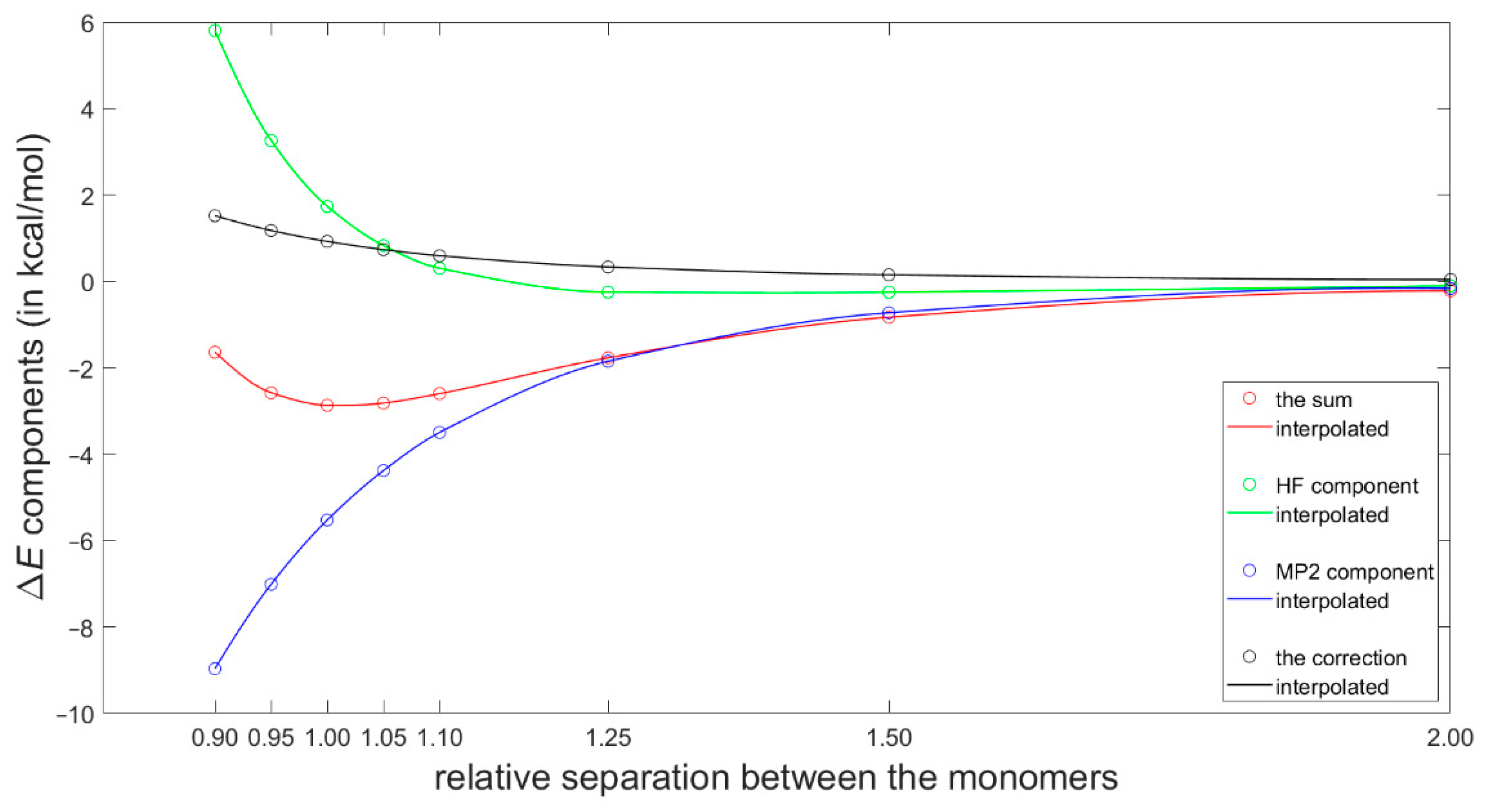
Most recently, Platzer et al. [5] discussed a possible connection between increasing binding enthalpy and a higher 1H CS perturbation for certain geometries of fragments involved in CH–π interactions in protein–ligand complexes. Specifically, they scanned the PES of a T-shaped benzene dimer using the ωB97X-D/cc-pVTZ dispersion corrected DFT approach (see Materials and Methods for referencing of this and the subsequent computational techniques), which had been previously shown to reliably describe the sandwich configuration [46]. Of practical significance is the fact that the ωB97X-D/cc-pVTZ interaction energy can be obtained using a tiny fraction of computational resources needed to estimate the corresponding “gold standard” coupled cluster singles and doubles with iterative inclusion of triples (CCSD(T))/complete basis set (CBS) value (such a value would at present be inaccessible for systems containing more than about 60 atoms) [47]. Moreover, the ωB97X-D method combined with a midsized basis set have recently been used in important investigations of stacking [16,48]. Hence, the ωB97X-D/cc-pVTZ approach was applied here to calculate interaction energies at the grid points for the 1H CS calculations described above. These interaction energies are provided in Table S4 and graphically presented in Figure S1. They are assumed to be quite accurate based on aforementioned references and also based on a high quality of the data obtained for the dissociation curve of the tilted T-shaped benzene dimer (see Figure 4 and Table S3). Namely, for its geometries reported in [29] and deposited in the BEGDB database [30], the ωB97X-D/cc-pVTZ results were compared to fully reliable CCSD(T)/CBS data and to their DFT-based symmetry-adapted intermolecular perturbation theory (DFT-SAPT)/CBS counterparts. Figure 4 shows only insignificant differences between the ωB97X-D/cc-pVTZ and the two sets of CBS-extrapolated values. The outstanding performance of the DFT-SAPT computational protocol, specified in Section 4, and whose results closely match the CCSD(T)/CBS points of the investigated curve are also noteworthy. As for the CCSD(T)/CBS values, they were reconstructed according to the description provided in [29] in order to examine their breakdown into the , , and components (see Equation (3) in Section 4). This breakdown is shown in Figure 5 and illustrates a delicate balance of contributions to the interaction energy.
3. Discussion
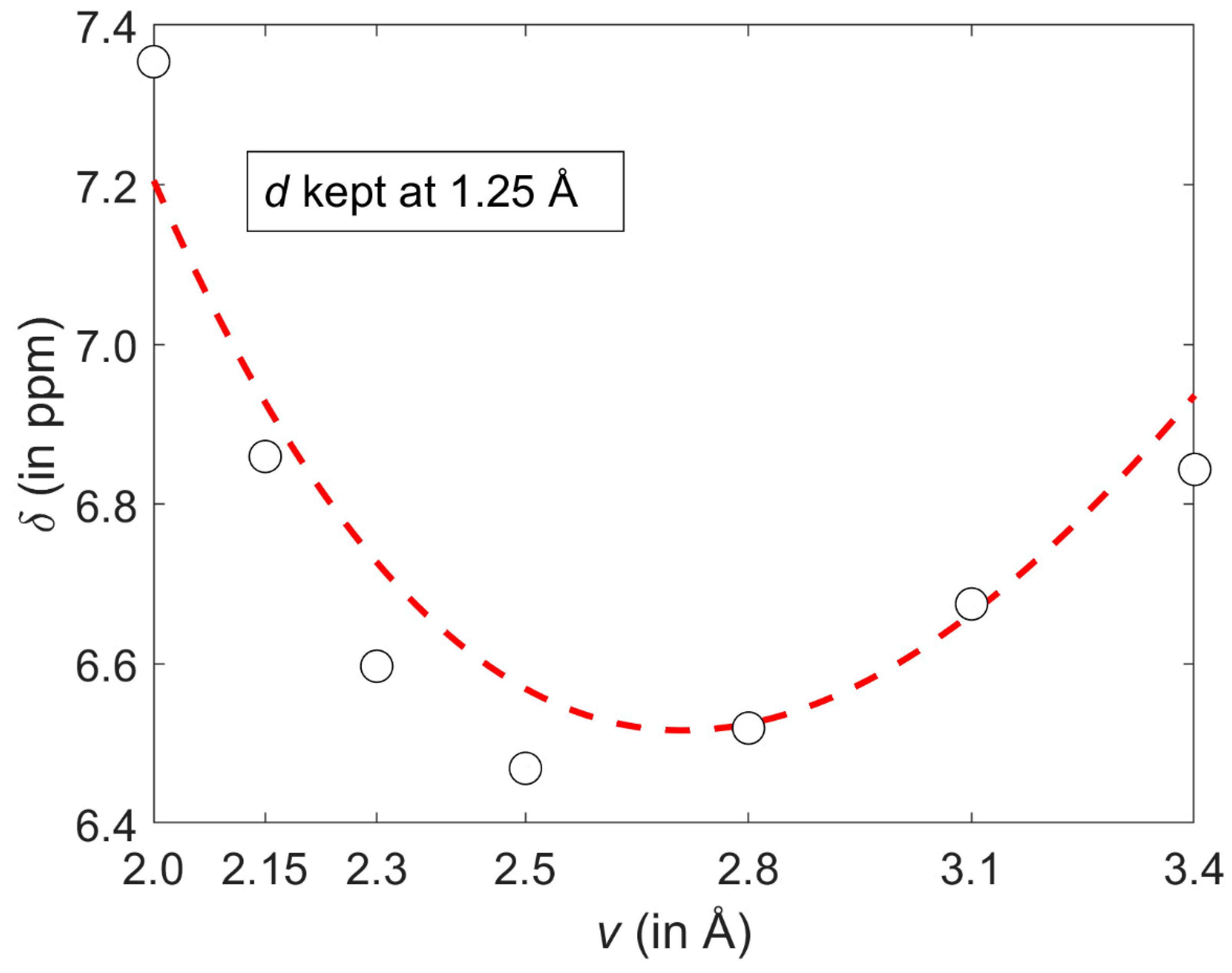
The GIAO-MP2 and GIAO-B3LYP methods were combined with the 6-311++G(2d,2p) basis set and applied to a relatively large interval of radial and angular orientations of the benzene dimer (both parametrizations are provided as Matlab m-files in Supplementary Materials). It is stressed that a majority of these orientations fulfil the commonly used criteria for aromatic C–H/ π bonding. Specifically, the Brandl–Weiss geometric system for identification of C–H/π interactions employs in a lateral dimension the coordinate denoted as [49] that is numerically equal to the coordinate d used here. In structural database searches, a cut-off value for of “1.0 or 1.2 Å for different sized π -acceptor systems“ is typically used [50] and would have covered most of the grid points if it had been applied in this work. Nevertheless, the GIAO calculations revealed a complicated landscape of the 1H CS values of the proton directly involved in the C–H/π interaction. These values do not change monotonically with increasing intermonomer separation for all lateral displacements considered here. An illustration of this non-monotonic behavior is provided in Figure 6 which shows a slice of the 1H CS surface taken at d = 1.25 Å. Using the parametrization expressed by Equation (1), a minimum of with respect to the vertical distance v can be easily obtained by taking the partial derivative and, after the (ρ, ϕ) → (v, d) coordinate transformation, solving for zero the right hand side of Equation (2) with d fixed at 1.25 Å.
At such a minimum point, the 1H CS is 6.5170 ppm and v is 2.7133 Å, apparently in agreement with Figure 6. For d = 1.25 Å and v = 2.7133 Å, the second derivative has a negative value (namely, −1.6529 ppm/Å2), of course confirming that the examined slice is convex. However, the 1H CS grows monotonically with increasing displacement in the whole interval of investigated vertical distances (an example of this dependence is given in Figure 7). As a consequence of completely different profiles in v and d dimensions, contour lines have an irregular shape, which would not be the case if simple models were applied (such models were carefully compared in a relatively recent study) [51]. Several contour lines are visualized in Figure 2, and numerical examples now follow. For the lowest vertical separation considered here (v = 2.002 Å), it is immediately found through Equation (1) that the particular values of 1H CS of [5.5, 6.0, and 6.5 ppm] are, respectively, reached at d = 0.7786, 0.9005, and 1.0305 Å. Then, using these displacement values, the 1H CS of 5.5, 6.0, and 6.5 ppm are located at v = 2.3648, 2.7317, and 3.0944 Å, respectively. This analysis explicitly shows that the same 1H CS value may occur in vastly different spatial arrangements of stacked molecules. Additional information would obviously be needed to resolve such ambiguities during a structure determination process, analogously to the requirement for neutron diffraction data in distinguishing hydrogen bond networks of some polymorphs [52].
4. Materials and Methods
The standard second-order Møller–Plesset (MP2) approach and the standard Becke’s three-parameter, Lee-Yang–Parr (B3LYP) combination of DFT functionals were used. In the chemical shielding calculations, these methods were combined with the standard 6-311++G(2d,2p) basis set and with the GIAO strategy to overcome the gauge problem [53,54]. Interaction energies corrected for the basis set superposition error by the counterpoise (CP) scheme [55] were obtained using the empirically corrected ωB97X-D DFT functional [56] and the standard cc-pVTZ (correlation-consistent polarized valence triple-ζ) basis set. In the full geometry optimization of tetramethylsilane, the MP2 method was combined with the standard aug-cc-pVTZ (the cc-pVTZ augmented with diffuse functions) basis set, and Td symmetry was imposed. All these calculations were carried out using the Gaussian 09 suite of codes [57].
Crystal structures [12,13] served as input for periodic DFT calculations which adopt the pseudopotential scheme [58,59,60] implemented in the CASTEP 16.1 program [60]. Unit-cell parameters of these structures were kept at experimental values, while atomic positions were optimized with respect to the crystal-lattice energy that was approximated using the Perdew–Burke–Erzerhof (PBE) DFT functional [61]. For resulting geometries, the chemical shielding was predicted by applying the GIPAW method [62,63] and the PBE functional. In all CASTEP calculations, the settings were consistent with “Fine” accuracy level of the Materials Studio 2019 software [64]. In particular, the PW cut-off value was 42.0 Ry. The ultrasoft on-the-fly generated pseudopotentials were adopted [65].
The CCSD(T) (coupled cluster singles and doubles with iterative inclusion of triples) interaction energies extrapolated to the CBS (complete basis set) limit, , were estimated using Equation (3)
The term is the interaction energy obtained at the Hartree–Fock (HF) level using the aug-cc-pVQZ basis set. The denotes the MP2 correlation energy contribution, which was obtained using the aug-cc-pVTZ and aug-cc-pVQZ values by their extrapolation that employed the scheme of Halkier et al. (Equation (7) in [66]). The component, which aims at correctly approximating higher-order correlation energy contributions to the interaction energy, was computed using the aug-cc-pVDZ basis set in the underlying CCSD(T) and MP2 calculations. The CP correction was applied throughout.
The DFT-based SAPT (symmetry-adapted intermolecular perturbation theory) [67] calculations (abbreviated as DFT-SAPT) were performed in the density-fitting variant [68]. The procedure described in [69] was followed except for an extrapolation of the interaction energy components. Namely, in this work all the components were extrapolated to their CBS limit and summed up, while Hesselmann et al. [69] extrapolated only the second-order dispersion contributions. The Molpro version 2008.1 [70] was used to obtain energies leading to the CCSD(T)/CBS and DFT-SAPT/CBS results.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/21/21/7908/s1, Table S1: Values of parameters in Equation (1), Table S2: The 1H chemical shielding data for the tilted T-shaped dimer of benzene, Table S3: Interaction energies of the tilted T-shaped dimer of benzene, Table S4: Predicted values of the 1H CS and of the ωB97X-D/cc-pVTZ interaction energy at 49 grid points, Figure S1: A plot of the ωB97X-D/cc-pVTZ interaction energies, Figure S2: A plot of measured and computed 1H chemical shifts of selected protons in two molecular crystals; script files ‘f_b3lyp.m’ and ‘f_mp2.m’ to obtain an estimate of the 1H CS on parametrized δ (v, d) surface.
Author Contributions
Conceptualization, J.C. and J.B.; Investigation, writing, J.C.; Validation, funding acquisition, J.B. All authors have read and approved the final manuscript.
Funding
Ministry of Education, Youth and Sports of the Czech Republic project LTAUSA18011.
Acknowledgments
Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures. J.C. is grateful for the excellent support he has been receiving from the staff of the MetaCentrum VO (https://metavo.metacentrum.cz/).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Larive, C.K.; Barding, G.A.; Dinges, M.N. NMR spectroscopy for metabolomics and metabolic profiling. Anal. Chem. 2015, 87, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Mroue, K.H.; Sun, P.; Ramamoorthy, A. High-Resolution Proton NMR Spectroscopy of Polymers and Biological Solids. In Modern Magnetic Resonance, 2nd ed.; Webb, G.A., Ed.; Springer: Cham, Switzerland, 2018; pp. 521–536. [Google Scholar]
- Stone, A.J. The Theory of Intermolecular Forces, 1st ed.; Clarendon Press: Oxford, UK, 2002; pp. 56–63. [Google Scholar]
- Kudisch, B.; Maiuri, M.; Moretti, L.; Oviedo, M.B.; Wang, L.; Oblinsky, D.G.; Prud’homme, R.K.; Wong, B.M.; McGill, S.A.; Scholes, G.D. Ring currents modulate optoelectronic properties of aromatic chromophores at 25 T. Proc. Natl. Acad. Sci. USA 2020, 117, 11289–11298. [Google Scholar] [CrossRef] [PubMed]
- Platzer, G.; Mayer, M.; Beier, A.; Brüschweiler, S.; Fuchs, J.E.; Engelhardt, H.; Geist, L.; Bader, G.; Schörghuber, J.; Lichtenecker, R.; et al. PI by NMR: Probing CH–π Interactions in Protein–Ligand Complexes by NMR Spectroscopy. Angew. Chem. Int. Ed. 2020, 59, 14861–14868. [Google Scholar] [CrossRef] [PubMed]
- Rickhaus, M.; Jirasek, M.; Tejerina, L.; Gotfredsen, H.; Peeks, M.D.; Haver, R.; Jiang, H.-W.; Claridge, T.D.W.; Anderson, H.L. Global aromaticity at the nanoscale. Nat. Chem. 2020, 12, 236–241. [Google Scholar] [CrossRef]
- Gabryelczyk, B.; Cai, H.; Shi, X.; Sun, Y.; Swinkels, P.J.M.; Salentinig, S.; Pervushin, K.; Miserez, A. Hydrogen bond guidance and aromatic stacking drive liquid-liquid phase separation of intrinsically disordered histidine-rich peptides. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, S.R.; Griffin, J.M.; Broch, K.; Lesage, A.; Lemaur, V.; Dudenko, D.; Olivier, Y.; Sirringhaus, H.; Emsley, L.; Grey, C.P. Donor–acceptor stacking arrangements in bulk and thin-film high-mobility conjugated polymers characterized using molecular modelling and MAS and surface-enhanced solid-state NMR spectroscopy. Chem. Sci. 2017, 8, 3126–3136. [Google Scholar] [CrossRef] [Green Version]
- Bass, T.M.; Carr, C.R.; Sherbow, T.J.; Fettinger, J.C.; Berben, L.A. Syntheses of Square Planar Galluim Complexes and a Proton NMR Correlation Probing Metalloaromaticity. Inor. Chem. 2020, 59, 13517–13523. [Google Scholar] [CrossRef]
- Lampkin, B.J.; Karadakov, P.B.; VanVeller, B. Detailed Visualization of Aromaticity Using Isotropic Magnetic Shielding. Angew. Chem. Int. Ed. 2020, 59, 2–9. [Google Scholar] [CrossRef]
- Kilymis, D.; Bartók, A.P.; Pickard, C.J.; Forse, A.C.; Merlet, C. Efficient prediction of nucleus independent chemical shifts for polycyclic aromatic hydrocarbons. Phys. Chem. Chem. Phys. 2020, 22, 13746–13755. [Google Scholar] [CrossRef]
- Pöppler, A.-C.; Corlett, E.K.; Pearce, H.; Seymour, M.P.; Reid, M.; Montgomery, M.G.; Brown, S.P. Single-crystal X-ray diffraction and NMR crystallography of a 1:1 cocrystal of dithianon and pyrimethanil. Acta Cryst. C 2017, 73, 149–156. [Google Scholar] [CrossRef]
- Schwartz, E.; Lim, E.; Gowda, C.M.; Liscio, A.; Fenwick, O.; Tu, G.; Palermo, V.; de Gelder, R.; Cornelissen, J.J.L.M.; Van Eck, E.R.H.; et al. Synthesis, Characterization, and Surface Initiated Polymerization of Carbazole Functionalized Isocyanides. Chem. Mater. 2010, 8, 2597–2607. [Google Scholar] [CrossRef]
- Gowda, C.M.; Vasconcelos, F.; Schwartz, E.; Van Eck, E.R.H.; Marsman, M.; Cornelissen, J.J.L.M.; Rowan, A.E.; De Wijs, G.A.; Kentgens, A.P.M. Hydrogen bonding and chemical shifts assignments in carbazole functionalized isocyanides from solid-state NMR and first-principles calculations. Phys. Chem. Chem. Phys. 2011, 13, 13082–13095. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azais, T.; Asbrook, S.E.; Griffin, J.M.; Yates, J.R.; Pickard, J.C. First-Principles Calculation of NMR Parameters Using the Gauge Including Projector Augmented Wave Method: A Chemist’s Point of View. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, A.N.; Doney, A.C.; Wheeler, S.E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chain. J. Am. Chem. Soc. 2019, 141, 11027–11035. [Google Scholar] [CrossRef]
- D’Ischia, M.; Napolitano, A.; Pezzella, A.; Meredith, P.; Buehler, M. Melanin biopolymers: Tailoring chemical complexity for materials design. Angew. Chem. Int. Ed. 2020, 59, 11196–11205. [Google Scholar] [CrossRef]
- Lyu, Q.; Hsueh, N.; Chai, C.L.L. Unravelling the polydopamine mystery: Is the end in sight? Polym. Chem. 2019, 10, 5771–5777. [Google Scholar] [CrossRef]
- Proks, V.; Brus, J.; Pop-Georgievski, O.; Večerníková, E.; Wiśniewski, W.; Kotek, J.; Urbanová, M.; Rypáček, F. Thermal-Induced Transformation of Polydopamine Structures: An Efficient Route for the Stabilization of the Polydopamine Surfaces. Macromol. Chem. Phys. 2013, 214, 499–507. [Google Scholar] [CrossRef]
- Circu, M.; Filip, C. Closer to the polydopamine structure: New insights from a combined 13C/1H/2H solid-state NMR study on deuterated samples. Polym. Chem. 2018, 9, 3379–3387. [Google Scholar] [CrossRef]
- Loeffler, J.R.; Fernández-Quintero, M.L.; Schauperl, M.; Liedl, K.R. STACKED – Solvation Theory of Aromatic Complexes as Key for Estimating Drug Binding. J. Chem. Inf. Model. 2020, 60, 2304–2313. [Google Scholar] [CrossRef] [Green Version]
- Bartolomei, M.; Pirani, F.; Marques, J.M.C. Low-energy structures of benzene clusters with a novel accurate potential surface. J. Comput. Chem. 2015, 36, 2291–2301. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.R.; McDonald, A.R.; DePrince, A.E., III; Marshall, M.S.; Podeszwa, R.; Sherrill, C.D. Resolving the three-body contribution to the lattice energy of crystalline benzene: Benchmark results from coupled-cluster theory. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef]
- Miliordos, E.; Apra, E.; Xantheas, S.S. Benchmark Theoretical Study of the π−π Binding Energy in the Benzene Dimer. J. Phys. Chem. A 2014, 118, 7568–7578. [Google Scholar] [CrossRef]
- DiStasio, R.A., Jr.; Von Helden, G.; Steele, R.P.; Head-Gordon, M. On the T-shaped structures of the benzene dimer. Chem. Phys. Lett. 2007, 437, 277–283. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Exploring Accuracy Limits of Predictions of the 1H NMR Chemical Shielding Anisotropy in the Solid State. Molecules 2019, 24, 1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czernek, J.; Brus, J. Monitoring the Site-Specific Solid-State NMR Data in Oligopeptides. Int. J. Mol. Sci. 2020, 21, 2700. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Polymorphic Forms of Valinomycin Investigated by NMR Crystallography. Int. J. Mol. Sci. 2020, 21, 4907. [Google Scholar] [CrossRef]
- Řezáč, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef]
- Řezáč, J.; Jurečka, P.; Riley, K.E.; Černý, J.; Valdes, H.; Pluháčková, K.; Berka, K.; Řezáč, T.; Pitoňák, M.; Vondrášek, J.; et al. Quantum Chemical Benchmark Energy and Geometry Database for Molecular Clusters and Complex Molecular Systems (www.begdb.com): A Users Manual and Examples. Collect. Czech. Chem. Commun. 2008, 73, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.P. Applications of high-resolution 1H solid-state NMR. Solid State Nucl. Magn. Reson. 2012, 41, 1–27. [Google Scholar] [CrossRef]
- Dudenko, D.V.; Yates, J.R.; Harris, K.D.M.; Brown, S.P. An NMR crystallography DFT-D approach to analyse the role of intermolecular hydrogen bonding and π–π interactions in driving cocrystallisation of indomethacin and nicotinamide. CrystEngComm 2013, 15, 8797–8807. [Google Scholar] [CrossRef]
- Dudenko, D.V.; Williams, P.A.; Hughes, C.E.; Antzutkin, O.N.; Velaga, S.P.; Brown, S.P.; Harris, K.D.M. Exploiting the Synergy of Powder X-ray Diffraction and Solid-State NMR Spectroscopy in Structure Determination of Organic Molecular Solids. J. Phys. Chem. C 2013, 117, 12258–12265. [Google Scholar] [CrossRef] [PubMed]
- Czernek, J.; Brus, J. The covariance of the differences between experimental and theoretical chemical shifts as an aid for assigning two-dimensional heteronuclear correlation solid-state NMR spectra. Chem. Phys. Lett. 2014, 608, 334–339. [Google Scholar] [CrossRef]
- Carignani, E.; Borsacchi, S.; Bradley, J.P.; Brown, S.P.; Geppi, M. Strong Intermolecular Ring Current Influence on 1H Chemical Shifts in Two Crystalline Forms of Naproxen: A Combined Solid-State NMR and DFT Study. J. Phys. Chem. C 2013, 117, 17731–17740. [Google Scholar] [CrossRef]
- Czernek, J. On the solid-state NMR spectra of naproxen. Chem. Phys. Lett. 2015, 619, 230–235. [Google Scholar] [CrossRef]
- Corlett, E.K.; Blade, H.; Hughes, L.P.; Sidebottom, P.J.; Walker, D.; Walton, R.I.; Brown, S.P. Investigating discrepancies between experimental solid-state NMR and GIPAW calculation: N=C–N 13C and OH⋯O 1H chemical shifts in pyridinium fumarates and their cocrystals. Solid State Nucl. Magn. Reson. 2020, 108. [Google Scholar] [CrossRef]
- Hušák, M.; Jegorov, A.; Rohlíček, J.; Fitch, A.; Czernek, J.; Kobera, L.; Brus, J. Determining the Crystal Structures of Peptide Analogs of Boronic Acid in the Absence of Single Crystals: Intricate Motifs of Ixazomib Citrate Revealed by XRPD Guided by ss-NMR. Cryst. Growth Des. 2018, 18, 3616–3625. [Google Scholar] [CrossRef]
- Harris, R.K.; Hodgkinson, P.; Zorin, V.; Dumez, J.N.; Elena-Herrmann, B.; Emsley, L.; Salager, E.; Stein, R.S. Computation and NMR crystallography of terbutaline sulfate. Magn. Reson. Chem. 2010, 48, S103–S112. [Google Scholar] [CrossRef]
- Kerr, H.E.; Softley, L.K.; Suresh, K.; Nangia, A.; Hodgkinson, P.; Radosavjlevic Evans, I. A furosemide–isonicotinamide cocrystal: An investigation of properties and extensive structural disorder. CrystEngComm 2015, 17, 6707–6715. [Google Scholar] [CrossRef] [Green Version]
- Frantsuzov, I.; Ford, S.J.; Radosavjlevic Evans, I.; Horsewill, A.J.; Trommsdorff, H.P.; Johnson, M.R. Measurement of Proton Tunneling in Short Hydrogen Bonds in Single Crystals of 3,5 Pyridinedicarboxylic Acid Using Nuclear Magnetic Resonance Spectroscopy. Phys. Rev. Lett. 2014, 113. [Google Scholar] [CrossRef] [Green Version]
- Dračínský, M.; Hodgkinson, P. A molecular dynamics study of the effects of fast molecular motions on solid-state NMR parameters. CrystEngComm 2013, 15, 8705–8712. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, Y.; Malon, M.; Potrzebowski, M.J.; Paluch, P.; Amoreux, J.P. Accurate NMR determination of C–H or N–H distances for unlabeled molecules. Solid State Nucl. Magn. Reson. 2016, 73, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Mroue, K.H.; Ramamoorthy, A. Proton-Based Ultrafast Magic Angle Spinning Solid-State NMR Spectroscopy. Acc. Chem. Res. 2017, 50, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Lu, X.; Xu, W.; Troup, G.M.; McNevin, M.J.; Nie, H.; Su, Y. Quantifying Pharmaceutical Formulations from Proton Detected Solid-State NMR under Ultrafast Magic Angle Spinning. J. Pharm. Sci. 2020, 109, 3045–3053. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.G.; Margreiter, M.A.; Fuchs, J.E.; Von Grefenstein, S.; Tuatermann, C.S.; Liedl, K.R.; Fox, T. Heteroaromatic π-Stacking Energy Landscapes. J. Chem. Inf. Model. 2014, 54, 1371–1379. [Google Scholar] [CrossRef]
- Gyevy-Nagy, L.; Kállay, M.; Nagy, P.R. Integral-Direct and Parallel Implementation of the CCSD(T) Method: Algorithmic Developments and Large-Scale Applications. J. Chem. Theory Comput. 2020, 16, 366–384. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Doney, A.C.; Wheeler, S.E. Tuning Stacking Interactions between Asp–Arg Salt Bridges and Heterocyclic Drug Fragments. J. Chem. Inf. Model. 2019, 59, 149–158. [Google Scholar] [CrossRef]
- Brandl, M.; Weiss, M.S.; Jabs, A.; Sühnel, J.; Hilgenfeld, R. C–H … π-Interaction in Proteins. J. Mol. Biol. 2001, 307, 357–377. [Google Scholar] [CrossRef]
- Nishiyo, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Chakrabarti, P. CH–π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef]
- Sahakyan, A.B.; Vendruscolo, M. Analysis of the Contributions of Ring Current and Electric Field Effects to the Chemical Shifts of RNA Bases. J. Phys. Chem. B 2013, 117, 1989–1998. [Google Scholar] [CrossRef]
- Widdifield, C.M.; Farrell, J.D.; Cole, J.C.; Howard, J.A.K.; Hodgkinson, P. Resolving alternative organic crystal structures using density functional theory and NMR chemical shifts. Chem. Sci. 2020, 11, 2987–2992. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Frish, M.J.; Trucks, J.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First principles simulation: Ideas, illustrations, and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculations of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76. [Google Scholar] [CrossRef]
- BIOVIA Materials Studio. Dassault Systèmes, Vélizy-Villacoublay: Paris, France. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 22 September 2020).
- Gao, S.-P.; Pickard, C.J.; Perlov, A.; Milman, V. Core-Level Spectroscopy Calculation and the Plane Wave Pseudopotential Method. J. Phys. Condens. Matter 2009, 21. [Google Scholar] [CrossRef] [PubMed]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-set convergence in correlated calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. Wiley Interdiscip. Rev. Comput. Mol. Phys. 2019, 10. [Google Scholar] [CrossRef]
- Hesselmann, A.; Jansen, G.; Schütz, M. Density-functional theory-symmetry-adapted intermolecular perturbation theory with density fitting: A new efficient method to study intermolecular interaction energies. J. Chem. Phys. 2005, 122. [Google Scholar] [CrossRef]
- Hesselmann, A.; Jansen, G.; Schütz, M. Interaction Energy Contributions of H-Bonded and Stacked Structures of the AT and GC DNA Base Pairs from the Combined Density Functional Theoryand Intermolecular Perturbation Theory Approach. J. Am. Chem. Soc. 2006, 128, 11730–11731. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Hesselmann, A.; Kats, D.; Kohn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of the benzene dimer together with the Cartesian (v, d) and polar (ρ, ϕ) coordinate systems used in this work.
Figure 1.
Schematic representation of the benzene dimer together with the Cartesian (v, d) and polar (ρ, ϕ) coordinate systems used in this work.

Figure 2.
The geometry dependence of the GIAO-MP2/6-311++G(2d,2p) predicted 1H chemical shifts (magenta circles) and their parametrization (cyan stars are shown at the grid points) obtained for the T-shaped benzene dimer.
Figure 2.
The geometry dependence of the GIAO-MP2/6-311++G(2d,2p) predicted 1H chemical shifts (magenta circles) and their parametrization (cyan stars are shown at the grid points) obtained for the T-shaped benzene dimer.

Figure 3.
Comparison between theory and experiment for the proton solid-state nuclear magnetic resonance (1H SSNMR) parameters of two molecular crystals specified in the text. Green squares, blue circles, and red crosses pertain to the GIPAW-PBE, GIAO-B3LYP, and parametrized data, respectively (the green dashed line is y = −1.292*x + 31.56 ppm, while the blue straight line is y = −1.054*x + 31.60 ppm).
Figure 3.
Comparison between theory and experiment for the proton solid-state nuclear magnetic resonance (1H SSNMR) parameters of two molecular crystals specified in the text. Green squares, blue circles, and red crosses pertain to the GIPAW-PBE, GIAO-B3LYP, and parametrized data, respectively (the green dashed line is y = −1.292*x + 31.56 ppm, while the blue straight line is y = −1.054*x + 31.60 ppm).

Figure 4.
Binding energies of the tilted T-shaped benzene dimer described in the text.

Figure 5.
The CCSD(T)/CBS interaction energy components at points along the dissociation curve of the tilted T-shaped benzene dimer.
Figure 5.
The CCSD(T)/CBS interaction energy components at points along the dissociation curve of the tilted T-shaped benzene dimer.

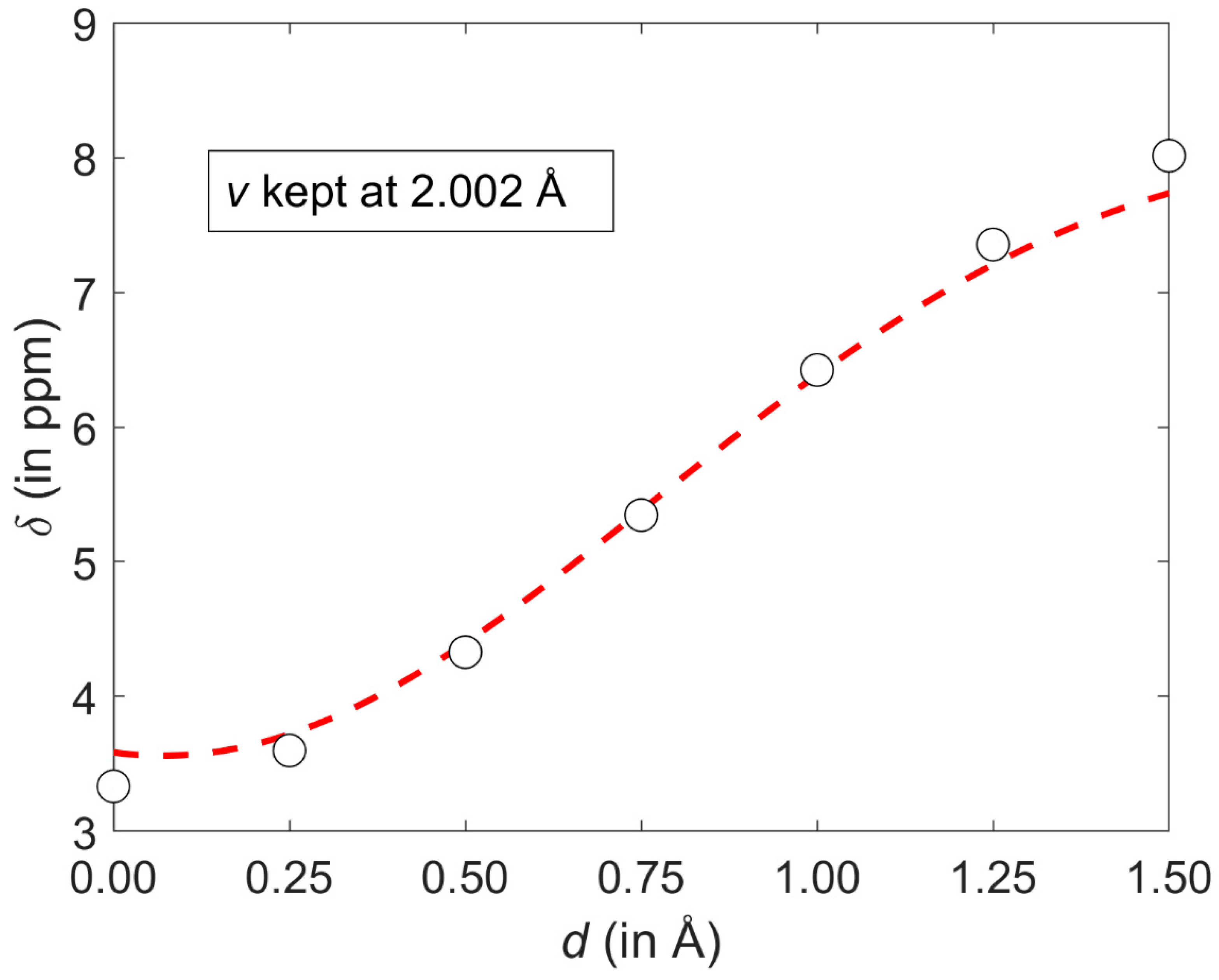
Figure 6.
A cut through the GIAO-MP2/6-311++G(2d,2p) 1H chemical shift (CS) surface discussed in the text. Values obtained from regular calculations and from the corresponding parametrization are shown as open black circles and the dashed red line, respectively.
Figure 6.
A cut through the GIAO-MP2/6-311++G(2d,2p) 1H chemical shift (CS) surface discussed in the text. Values obtained from regular calculations and from the corresponding parametrization are shown as open black circles and the dashed red line, respectively.

Figure 7.
A cut through the GIAO-MP2/6-311++G(2d,2p) 1H CS surface discussed in the text. Values obtained from regular calculations and from the corresponding parametrization are shown as open black circles and the dashed red line, respectively.
Figure 7.
A cut through the GIAO-MP2/6-311++G(2d,2p) 1H CS surface discussed in the text. Values obtained from regular calculations and from the corresponding parametrization are shown as open black circles and the dashed red line, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Structural and spectral parameters of investigated solid-phase systems. In this table, the measured chemical shift is denoted as δ, and its theoretical estimate as δ’. See the text for details.
Table 1.
Structural and spectral parameters of investigated solid-phase systems. In this table, the measured chemical shift is denoted as δ, and its theoretical estimate as δ’. See the text for details.
| Crystal | Site | v (in Å) | d (in Å) | δ (in ppm) | σ/δ’ Periodic (in ppm) | σ/δ’ Cluster (in ppm) | σ/δ’ Fit (in ppm) |
|---|---|---|---|---|---|---|---|
| Dithianon–pyrimethanil | H25 | 2.5238 | 0.5056 | 4.0 | 26.9417/3.9 | 27.2285/4.6 | 26.5599/5.3 |
| H2 | 2.6975 | 0.9521 | 6.2 | 24.4601/6.4 | 24.8389/7.0 | 25.6178/6.2 | |
| The isocyanide | H10′ | 2.6009 | 0.1846 | 4.8 | 26.2797/4.6 | 26.7231/5.1 | 26.7550/5.1 |
| H11′ | 2.7557 | 0.2720 | 5.8 | 25.0655/5.8 | 25.6729/6.2 | 26.4137/5.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Czernek, J.; Brus, J. Parametrizing the Spatial Dependence of 1H NMR Chemical Shifts in π-Stacked Molecular Fragments. Int. J. Mol. Sci. 2020, 21, 7908. https://doi.org/10.3390/ijms21217908
AMA Style
Czernek J, Brus J. Parametrizing the Spatial Dependence of 1H NMR Chemical Shifts in π-Stacked Molecular Fragments. International Journal of Molecular Sciences. 2020; 21(21):7908. https://doi.org/10.3390/ijms21217908
Chicago/Turabian StyleCzernek, Jiří, and Jiří Brus. 2020. "Parametrizing the Spatial Dependence of 1H NMR Chemical Shifts in π-Stacked Molecular Fragments" International Journal of Molecular Sciences 21, no. 21: 7908. https://doi.org/10.3390/ijms21217908
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.