Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Weight Parameters and Heart Function
2.2. Necroptosis Signaling
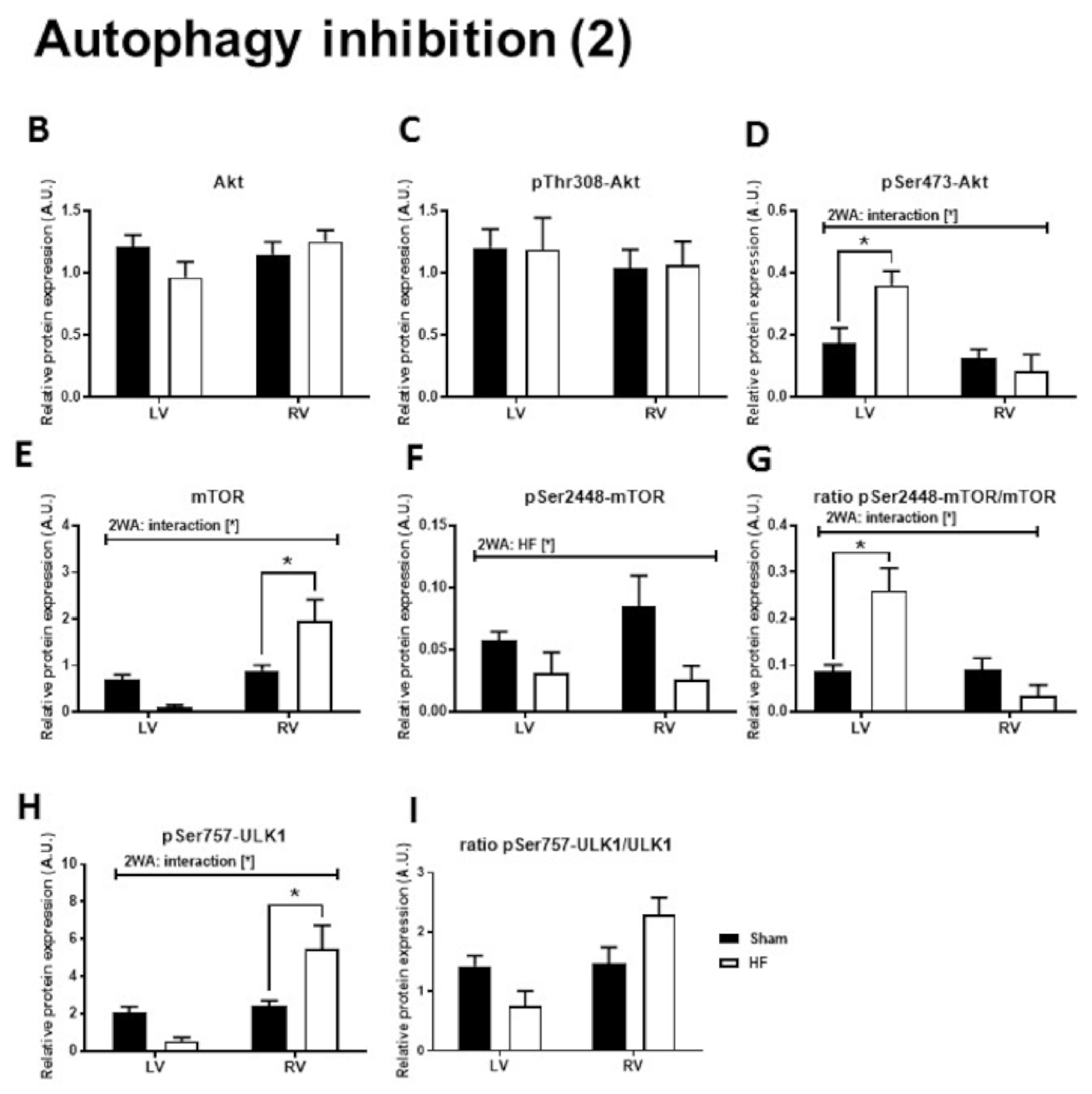
2.3. Autophagy Signaling
2.4. Apoptosis Signaling
3. Discussion
4. Materials and Methods
4.1. Experimental Groups
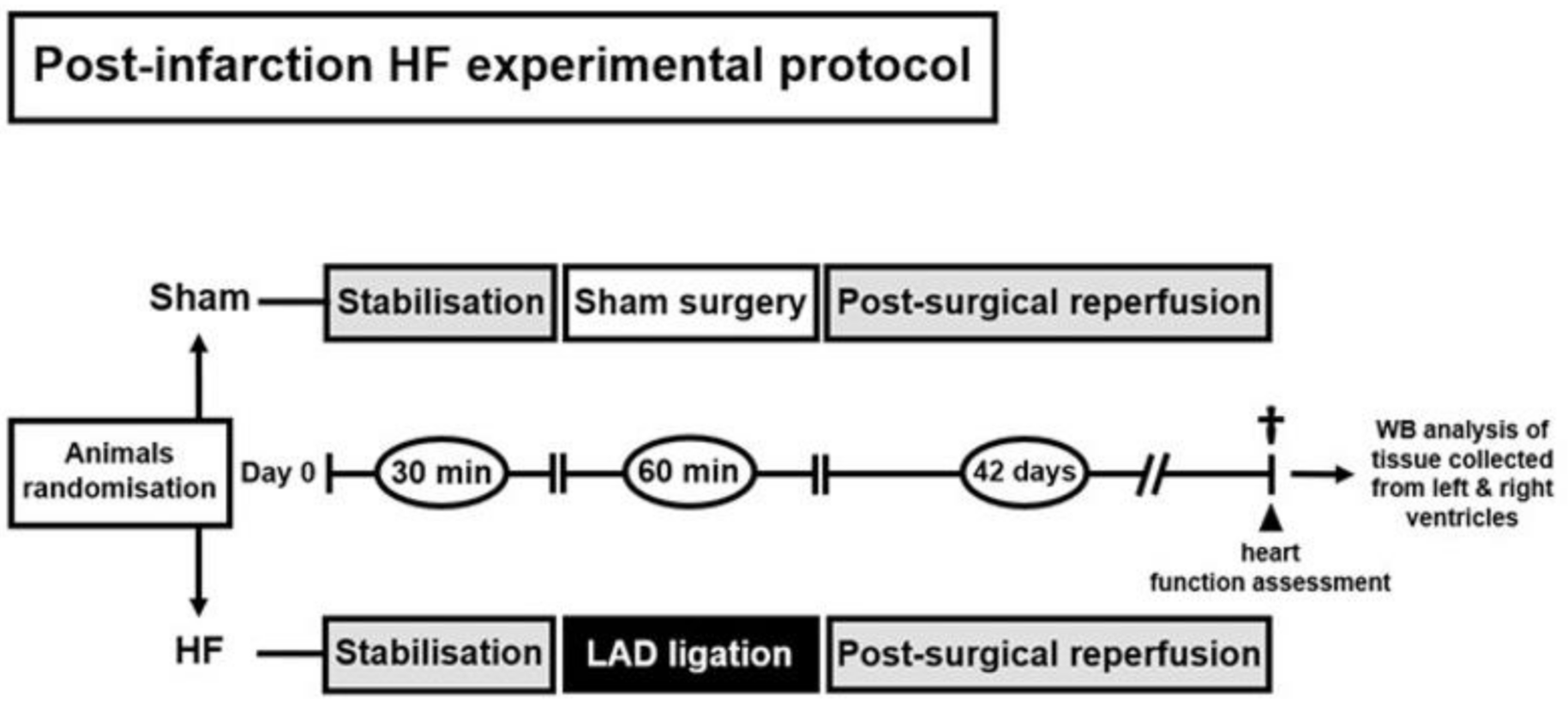
4.2. Animal Model of Post-Myocardial Infarction Heart Failure and Study Design
4.3. Invasive Cardiac Function Assessment
4.4. SDS-PAGE and Immunoblotting
4.5. Statistical Analysis
5. Conclusions
6. Study limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pasumarthi, K.B.; Field, L.J. Cardiomyocyte cell cycle regulation. Circ. Res. 2002, 90, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arter. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, G.W.; Marin-Garcia, J. Role of cell death in the progression of heart failure. Heart Fail. Rev. 2016, 21, 157–167. [Google Scholar] [CrossRef]
- Corsetti, G.; Chen-Scarabelli, C.; Romano, C.; Pasini, E.; Dioguardi, F.S.; Onorati, F.; Knight, R.; Patel, H.; Saravolatz, L.; Faggian, G.; et al. Autophagy and Oncosis/Necroptosis Are Enhanced in Cardiomyocytes from Heart Failure Patients. Med. Sci. Monit. Basic Res. 2019, 25, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, P.; Wang, Y.; Du, Y.; Nan, A.; Liu, S.; Zhang, Y.; Zhou, N.; Xu, Z.; Yang, Z. Ad-HGF improves the cardiac remodeling of rat following myocardial infarction by upregulating autophagy and necroptosis and inhibiting apoptosis. Am. J. Transl. Res. 2016, 8, 4605–4627. [Google Scholar] [PubMed]
- Wang, X.; Guo, Z.; Ding, Z.; Mehta, J.L. Inflammation, Autophagy, and Apoptosis After Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e008024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szobi, A.; Goncalvesova, E.; Varga, Z.V.; Leszek, P.; Kusmierczyk, M.; Hulman, M.; Kyselovic, J.; Ferdinandy, P.; Adameova, A. Analysis of necroptotic proteins in failing human hearts. J. Transl. Med. 2017, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Lichy, M.; Szobi, A.; Hrdlicka, J.; Horvath, C.; Kormanova, V.; Rajtik, T.; Neckar, J.; Kolar, F.; Adameova, A. Different signalling in infarcted and non-infarcted areas of rat failing hearts: A role of necroptosis and inflammation. J. Cell Mol. Med. 2019, 23, 6429–6441. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, M.I.; Liu, J.; Arslan, F.; den Ouden, K.; van Middelaar, B.J.; Doevendans, P.A.; Sluijter, J.P. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol. 2012, 107, 270. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Z.; Li, L.; Zhong, C.Q.; Zheng, X.; Wu, X.; Zhang, Y.; Ma, H.; Huang, D.; Li, W.; et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 2013, 288, 16247–16261. [Google Scholar] [CrossRef] [Green Version]
- Ros, U.; Pena-Blanco, A.; Hanggi, K.; Kunzendorf, U.; Krautwald, S.; Wong, W.W.; Garcia-Saez, A.J. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep. 2017, 19, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, Z.G. Execution of RIPK3-regulated necrosis. Mol. Cell Oncol. 2014, 1, e960759. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.K.; Adameova, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.M.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H891–H922. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Cook, W.D.; Okamoto, T.; Murphy, J.; Lawlor, K.E.; Vince, J.E.; Vaux, D.L. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis. 2013, 4, e465. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Wu, J.; Dang, Y.; Su, W.; Liu, C.; Ma, H.; Shan, Y.; Pei, Y.; Wan, B.; Guo, J.; Yu, L. Molecular cloning and characterization of rat LC3A and LC3B--two novel markers of autophagosome. Biochem. Biophys. Res. Commun. 2006, 339, 437–442. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Nishida, K.; Kyoi, S.; Yamaguchi, O.; Sadoshima, J.; Otsu, K. The role of autophagy in the heart. Cell Death Differ. 2009, 16, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Adameova, A.; Goncalvesova, E.; Szobi, A.; Dhalla, N.S. Necroptotic cell death in failing heart: Relevance and proposed mechanisms. Heart Fail. Rev. 2016, 21, 213–221. [Google Scholar] [CrossRef]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, M.; Yano, T.; Tanno, M.; Abe, K.; Ishikawa, S.; Miki, T.; Kuno, A.; Tobisawa, T.; Muratsubaki, S.; Ohno, K.; et al. Suppression of autophagic flux contributes to cardiomyocyte death by activation of necroptotic pathways. J. Mol. Cell Cardiol. 2017, 108, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Sun, A. Programmed necrosis in heart disease: Molecular mechanisms and clinical implications. J. Mol. Cell Cardiol. 2018, 116, 125–134. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, G.R.; De Keulenaer, G.W.; Martinet, W. Role of autophagy in heart failure associated with aging. Heart Fail. Rev. 2010, 15, 423–430. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Zhang, C.; Li, J.; Guo, W.; Yan, D.; Yang, C.; Zhao, J.; Xia, T.; Wang, Y.; et al. Heat shock protein 70 inhibits cardiomyocyte necroptosis through repressing autophagy in myocardial ischemia/reperfusion injury. In Vitro Cell Dev. Biol. Anim. 2016, 52, 690–698. [Google Scholar] [CrossRef]
- Jose Corbalan, J.; Vatner, D.E.; Vatner, S.F. Myocardial apoptosis in heart disease: Does the emperor have clothes? Basic Res. Cardiol. 2016, 111, 31. [Google Scholar] [CrossRef]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stennicke, H.R.; Jurgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yin, Y.; Liu, Y.; Zou, G.; Huang, H.; Qian, P.; Zhang, G.; Zhang, J. Necroptosis Mediated by Impaired Autophagy Flux Contributes to Adverse Ventricular Remodeling after Myocardial Infarction. Biochem. Pharm. 2020, 175, 113915. [Google Scholar] [CrossRef] [PubMed]
- Hrdlicka, J.; Neckar, J.; Papousek, F.; Vasinova, J.; Alanova, P.; Kolar, F. Beneficial effect of continuous normobaric hypoxia on ventricular dilatation in rats with post-infarction heart failure. Physiol. Res. 2016, 65, 867–870. [Google Scholar] [CrossRef]
- Bai, X.; Jiang, Y. Key factors in mTOR regulation. Cell Mol. Life Sci. 2010, 67, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, V.C.; Markworth, J.F.; Cameron-Smith, D. Considerations on mTOR regulation at serine 2448: Implications for muscle metabolism studies. Cell Mol. Life Sci. 2017, 74, 2537–2545. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sin, K.W.T.; Ding, H.; Doan, H.A.; Gao, S.; Miao, H.; Wei, Y.; Wang, Y.; Zhang, G.; Li, Y.P. p38beta MAPK mediates ULK1-dependent induction of autophagy in skeletal muscle of tumor-bearing mice. Cell Stress 2018, 2, 311–324. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.C.; Yu, L.; Wang, H.J.; Tashiro, S.; Onodera, S.; Ikejima, T. TNFalpha-induced necroptosis and autophagy via supression of the p38-NF-kappaB survival pathway in L929 cells. J. Pharm. Sci. 2011, 117, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.W.; Vince, J.E.; Lalaoui, N.; Lawlor, K.E.; Chau, D.; Bankovacki, A.; Anderton, H.; Metcalf, D.; O’Reilly, L.; Jost, P.J.; et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 2014, 123, 2562–2572. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Li, J. Inflammation and Inflammatory Cells in Myocardial Infarction and Reperfusion Injury: A Double-Edged Sword. Clin. Med. Insights Cardiol. 2016, 10, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The immune system and the remodeling infarcted heart: Cell biological insights and therapeutic opportunities. J. Cardiovasc. Pharm. 2014, 63, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Kuranaga, E.; Miura, M. Nonapoptotic functions of caspases: Caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol. 2007, 17, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Communal, C.; Sumandea, M.; de Tombe, P.; Narula, J.; Solaro, R.J.; Hajjar, R.J. Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 6252–6256. [Google Scholar] [CrossRef] [Green Version]
- Cervenka, L.; Huskova, Z.; Kopkan, L.; Kikerlova, S.; Sedlakova, L.; Vanourkova, Z.; Alanova, P.; Kolar, F.; Hammock, B.D.; Hwang, S.H.; et al. Two pharmacological epoxyeicosatrienoic acid-enhancing therapies are effectively antihypertensive and reduce the severity of ischemic arrhythmias in rats with angiotensin II-dependent hypertension. J. Hypertens. 2018, 36, 1326–1341. [Google Scholar] [CrossRef]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharm. 2018, 175, 987–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckar, J.; Hye Khan, M.A.; Gross, G.J.; Cyprova, M.; Hrdlicka, J.; Kvasilova, A.; Falck, J.R.; Campbell, W.B.; Sedlakova, L.; Skutova, S.; et al. Epoxyeicosatrienoic acid analog EET-B attenuates post-myocardial infarction remodeling in spontaneously hypertensive rats. Clin. Sci. (Lond.) 2019, 133, 939–951. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham-Operated | Heart Failure | |
|---|---|---|
| BW (g) | 519 ± 22 | 510 ± 26 |
| HW (mg) | 1.40 ± 0.08 | 1.63 ± 0.09 |
| LW (mg) | 1.52 ± 0.05 | 3.78 ± 0.65 * |
| HW/BW (mg/g) | 2.69 ± 0.08 | 3.23 ± 0.24 * |
| LW/BW (mg/g) | 2.94 ± 0.08 | 7.53 ± 1.33 * |
| LVDevP (mm Hg) | 116 ± 5 | 87 ± 5 * |
| LVEDP (mm Hg) | 9.0 ± 1.5 | 32.1 ± 4.3 * |
| +(dP/dt)max (mm Hg/s) | 6349 ± 400 | 3990 ± 306 * |
| −(dP/dt)max (mm Hg/s) | 5613 ± 589 | 3127 ± 354 * |
| HR (beats/min) | 333 ± 13 | 318 ± 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lichý, M.; Szobi, A.; Hrdlička, J.; Neckář, J.; Kolář, F.; Adameová, A. Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure. Int. J. Mol. Sci. 2020, 21, 7782. https://doi.org/10.3390/ijms21207782
Lichý M, Szobi A, Hrdlička J, Neckář J, Kolář F, Adameová A. Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure. International Journal of Molecular Sciences. 2020; 21(20):7782. https://doi.org/10.3390/ijms21207782
Chicago/Turabian StyleLichý, Martin, Adrián Szobi, Jaroslav Hrdlička, Jan Neckář, František Kolář, and Adriana Adameová. 2020. "Programmed Cell Death in the Left and Right Ventricle of the Late Phase of Post-Infarction Heart Failure" International Journal of Molecular Sciences 21, no. 20: 7782. https://doi.org/10.3390/ijms21207782