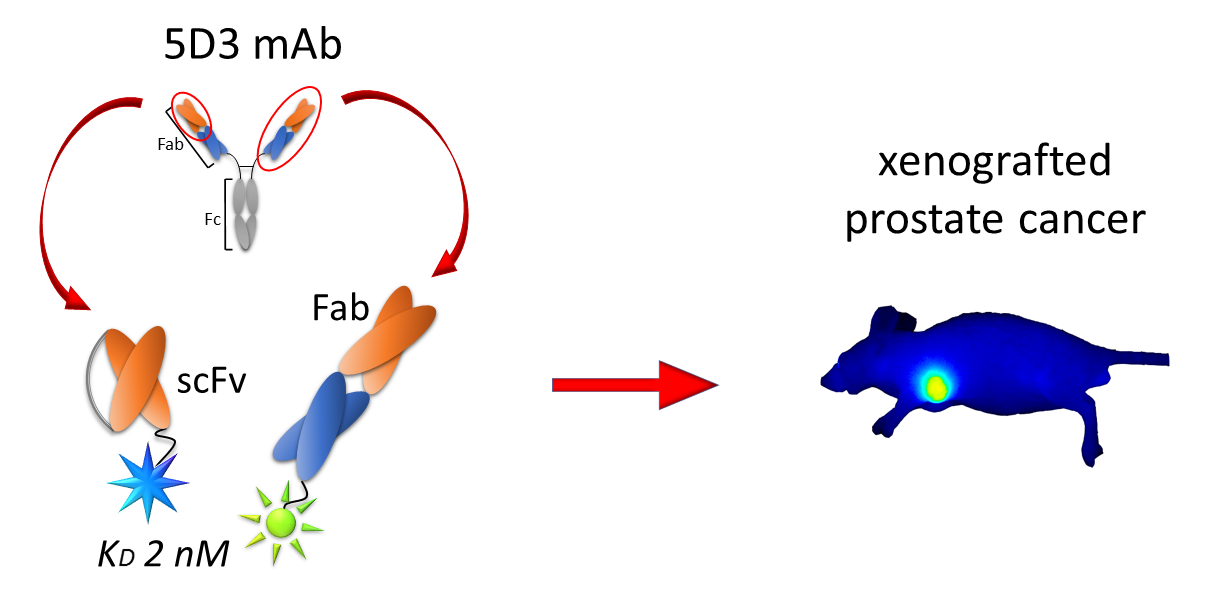
Engineered Fragments of the PSMA-Specific 5D3 Antibody and Their Functional Characterization

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results
2.1. Identification of Nucleotide and Amino Acid Sequences of the 5D3 mAb
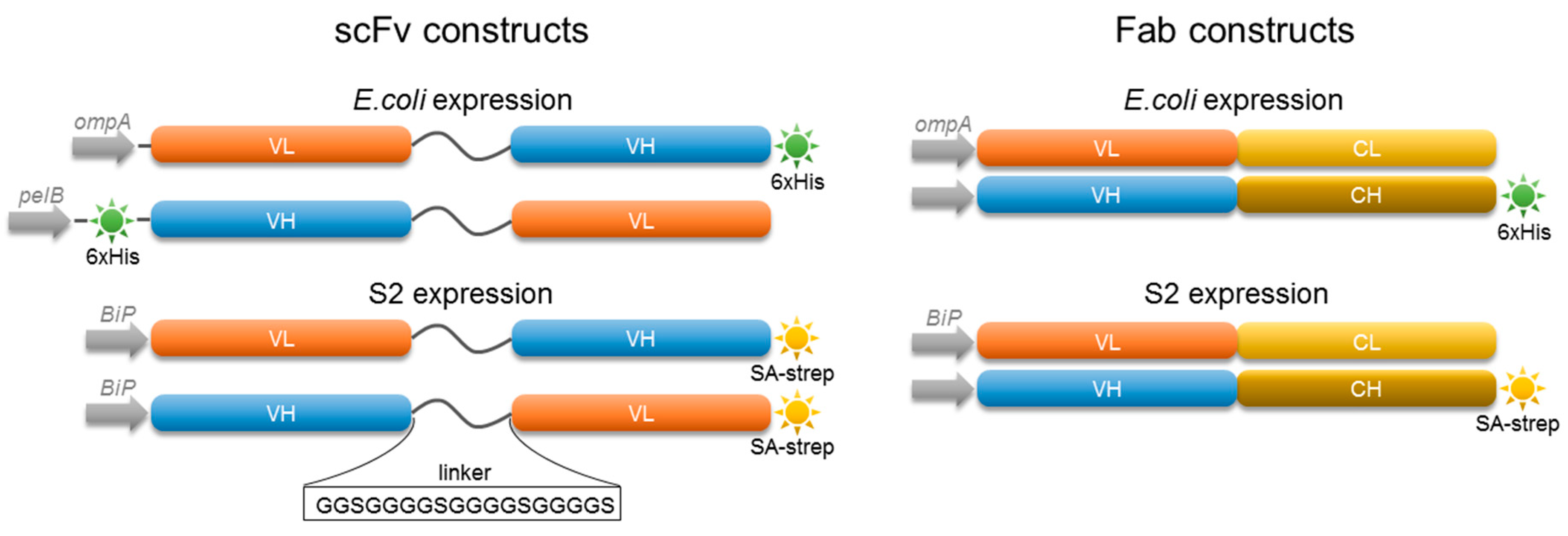
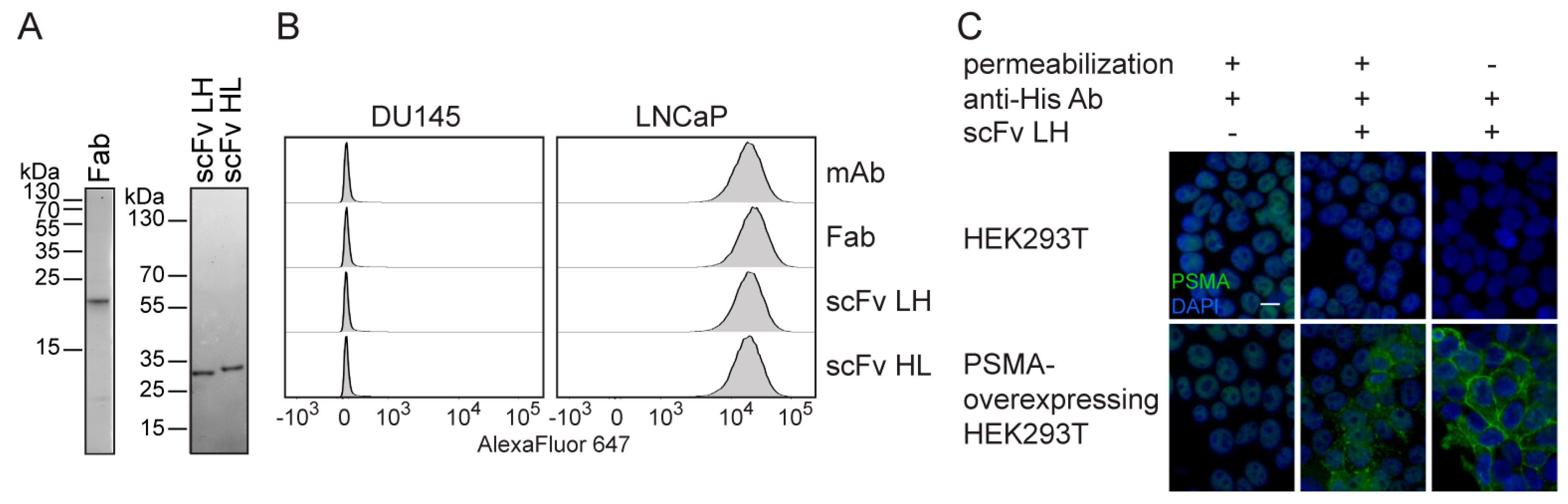
2.2. Characterization of 5D3 Antibody Fragments Produced in E. coli
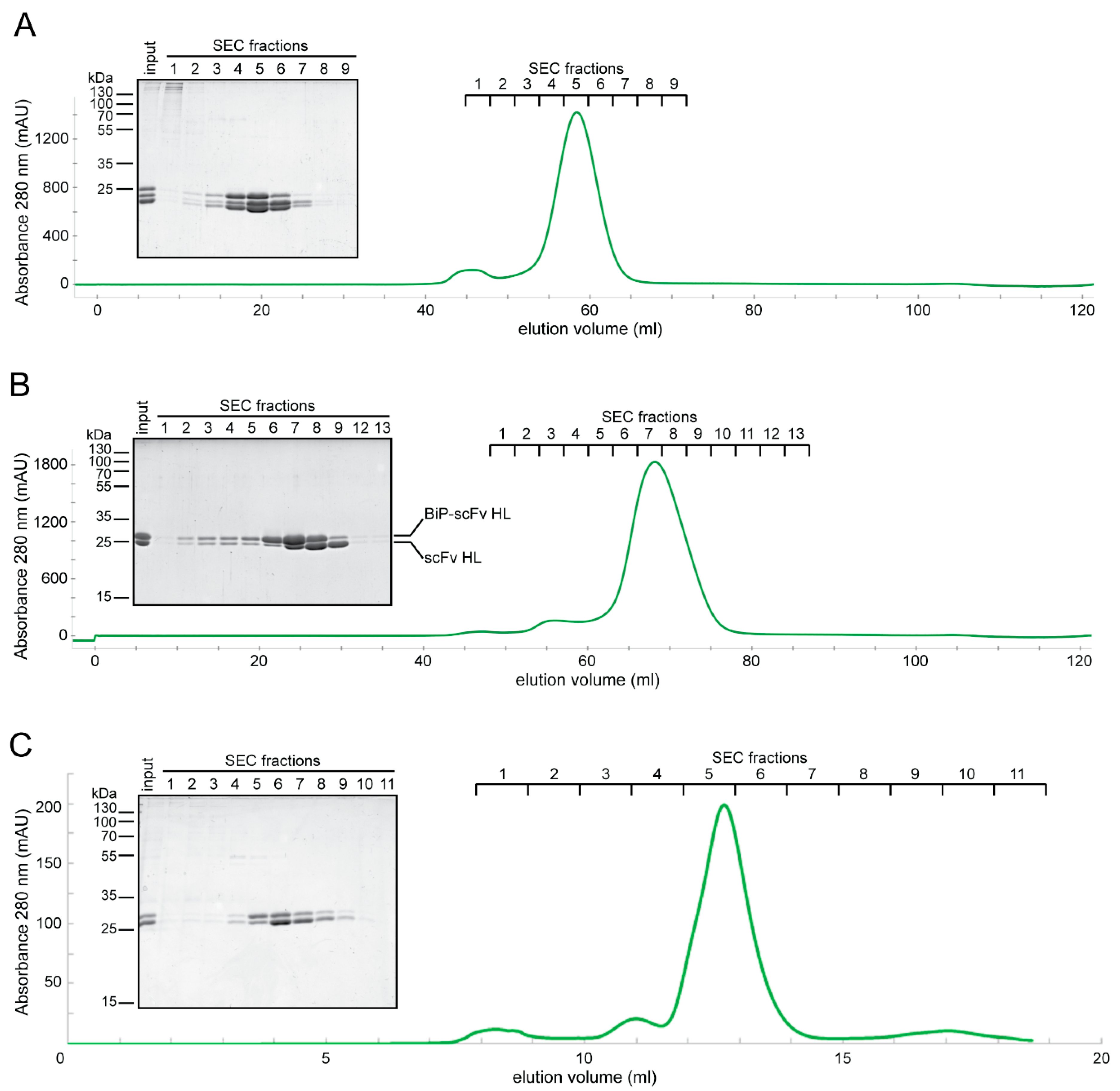
2.3. Expression of Fab and scFv Fragments in Insect S2 Cells
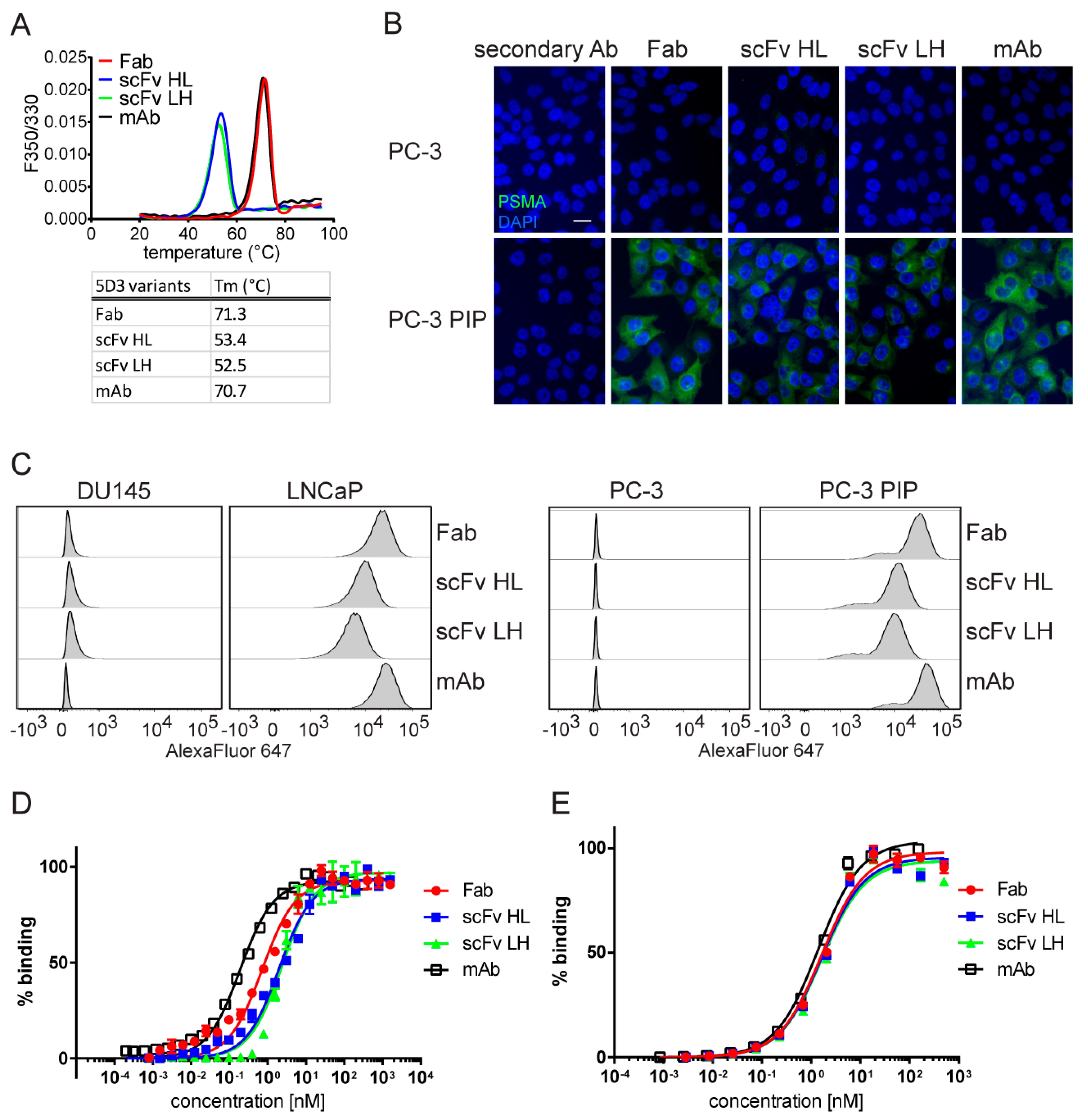
2.4. In Vitro Characterization of S2-Produced 5D3 Variants
2.5. In Vivo PSMA Imaging in an Experimental PCa Mouse Model
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. N-Terminal Amino Acid Sequencing
4.3. RNA Isolation and cDNA Synthesis
4.4. Antibody Cloning
4.5. Mass Spectrometry Analysis of the Antibody Sequence
4.6. Construction of Recombinant Antibody Expression Vectors
4.7. Cell Lines
4.8. Generation of Stably Transfected Insect Cells
4.9. Expression and Purification of Engineered Fragments Produced in E. coli
4.10. Expression and Purification of Engineered Fragments Produced in Insect Cells
4.11. Purification of PSMA Extracellular Domain
4.12. SDS Polyacrylamide Electrophoresis and Western Blotting
4.13. Indirect Immunofluorescence Microscopy
4.14. ELISA
4.15. Flow Cytometry
4.16. Nanoscale Differential Scanning Fluorimetry
4.17. Fluorescence Dye Conjugation
4.18. In Vivo Imaging Using Near-Infrared Fluorescence (NIRF)
4.19. Ex Vivo Imaging
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| appKD | Apparent dissociation constant |
| BSA | Bovine serum albumin |
| CBB | Coomassie Brilliant Blue G-250 |
| ELISA | Enzyme-linked immunosorbent assay |
| HL | Heavy-to-light |
| IF | Indirect immunofluorescence |
| LH | Light-to-heavy |
| mAb | Monoclonal antibody |
| MALDI-TOF | Matrix assisted laser desorption/ionization–time-of-flight |
| NAALADase | N acetylated-alpha-linked acidic dipeptidase |
| nanoDSF | Differential scanning fluorimetry |
| p.i. | Post-injection |
| PBS | Phosphate buffered saline |
| PCa | Prostate cancer |
| PSMA | Prostate-specific membrane antigen |
| PVDF | Polyvinylidene fluoride |
| SEC | Size exclusion chromatography |
| scFv | Single chain variable fragment |
| Tm | Melting temperature |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, T.; Eiber, M.; Schwaiger, M.; Gschwend, J.E. Current use of PSMA-PET in prostate cancer management. Nat. Rev. Urol. 2016, 13, 226–235. [Google Scholar] [CrossRef]
- Zhao, X.; Ning, Q.; Mo, Z.; Tang, S. A promising cancer diagnosis and treatment strategy: Targeted cancer therapy and imaging based on antibody fragment. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3621–3630. [Google Scholar] [CrossRef] [Green Version]
- Batra, S.K.; Jain, M.; Wittel, U.A.; Chauhan, S.C.; Colcher, D. Pharmacokinetics and biodistribution of genetically engineered antibodies. Curr. Opin. Biotechnol. 2002, 13, 603–608. [Google Scholar] [CrossRef]
- Bauerschlag, D.; Meinhold-Heerlein, I.; Maass, N.; Bleilevens, A.; Brautigam, K.; Al Rawashdeh, W.; Di Fiore, S.; Haugg, A.M.; Gremse, F.; Steitz, J.; et al. Detection and Specific Elimination of EGFR(+) Ovarian Cancer Cells Using a Near Infrared Photoimmunotheranostic Approach. Pharm. Res. 2017, 34, 696–703. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, M.; Chen, X.; Zhang, X.; Sukhadia, S.; Musolino, N.; Bao, H.; Chen, T.; Xu, C.; Wang, Q.; et al. Characterization of the first fully human anti-TEM1 scFv in models of solid tumor imaging and immunotoxin-based therapy. Cancer Immunol. Immunother. 2017, 66, 367–378. [Google Scholar] [CrossRef]
- Rabenhold, M.; Steiniger, F.; Fahr, A.; Kontermann, R.E.; Ruger, R. Bispecific single-chain diabody-immunoliposomes targeting endoglin (CD105) and fibroblast activation protein (FAP) simultaneously. J. Control. Release 2015, 201, 56–67. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Zhang, M.; Kobayashi, N.; Zettlitz, K.A.; Kono, E.A.; Yamashiro, J.M.; Tsai, W.K.; Jiang, Z.K.; Tran, C.P.; Wang, C.; Guan, J.; et al. Near-Infrared Dye-Labeled Anti-Prostate Stem Cell Antigen Minibody Enables Real-Time Fluorescence Imaging and Targeted Surgery in Translational Mouse Models. Clin. Cancer Res. 2019, 25, 188–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, S.M.; Tavare, R.; Zettlitz, K.A.; Rochefort, M.M.; Salazar, F.B.; Jiang, Z.K.; Reiter, R.E.; Wu, A.M. Applications of immunoPET: Using 124I-anti-PSCA A11 minibody for imaging disease progression and response to therapy in mouse xenograft models of prostate cancer. Clin. Cancer Res. 2014, 20, 6367–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonstra, M.C.; Tolner, B.; Schaafsma, B.E.; Boogerd, L.S.; Prevoo, H.A.; Bhavsar, G.; Kuppen, P.J.; Sier, C.F.; Bonsing, B.A.; Frangioni, J.V.; et al. Preclinical evaluation of a novel CEA-targeting near-infrared fluorescent tracer delineating colorectal and pancreatic tumors. Int. J. Cancer 2015, 137, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Debie, P.; Hernot, S. Emerging Fluorescent Molecular Tracers to Guide Intra-Operative Surgical Decision-Making. Front. Pharmacol. 2019, 10, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.L., Jr.; Haley, C.; Beckett, M.L.; Schellhammer, P.F. Expression of prostate-specific membrane antigen in normal, benign, and malignant prostate tissues. Urol. Oncol. 1995, 1, 18–28. [Google Scholar] [CrossRef]
- Frigerio, B.; Franssen, G.; Luison, E.; Satta, A.; Seregni, E.; Colombatti, M.; Fracasso, G.; Valdagni, R.; Mezzanzanica, D.; Boerman, O.; et al. Full preclinical validation of the 123I-labeled anti-PSMA antibody fragment ScFvD2B for prostate cancer imaging. Oncotarget 2017, 8, 10919–10930. [Google Scholar] [CrossRef] [Green Version]
- Czerwinska, M.; Bilewicz, A.; Kruszewski, M.; Wegierek-Ciuk, A.; Lankoff, A. Targeted Radionuclide Therapy of Prostate Cancer-From Basic Research to Clinical Perspectives. Molecules 2020, 25, 1743. [Google Scholar] [CrossRef] [Green Version]
- Foss, C.A.; Mease, R.C.; Cho, S.Y.; Kim, H.J.; Pomper, M.G. GCPII imaging and cancer. Curr. Med. Chem. 2012, 19, 1346–1359. [Google Scholar] [CrossRef]
- Barinka, C.; Rojas, C.; Slusher, B.; Pomper, M. Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr. Med. Chem. 2012, 19, 856–870. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, S.; Mullen, G.E.D.; Blower, P.J.; Ballinger, J.R. A 99mTc-labelled scFv antibody fragment that binds to prostate-specific membrane antigen. Nucl. Med. Commun. 2017, 38, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Mazzocco, C.; Fracasso, G.; Germain-Genevois, C.; Dugot-Senant, N.; Figini, M.; Colombatti, M.; Grenier, N.; Couillaud, F. In vivo imaging of prostate cancer using an anti-PSMA scFv fragment as a probe. Sci. Rep. 2016, 6, 23314. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, B.; Morlino, S.; Luison, E.; Seregni, E.; Lorenzoni, A.; Satta, A.; Valdagni, R.; Bogni, A.; Chiesa, C.; Mira, M.; et al. Anti-PSMA (124)I-scFvD2B as a new immuno-PET tool for prostate cancer: Preclinical proof of principle. J. Exp. Clin. Cancer Res. 2019, 38, 326. [Google Scholar] [CrossRef] [PubMed]
- Lutje, S.; van Rij, C.M.; Franssen, G.M.; Fracasso, G.; Helfrich, W.; Eek, A.; Oyen, W.J.; Colombatti, M.; Boerman, O.C. Targeting human prostate cancer with 111In-labeled D2B IgG, F(ab’)2 and Fab fragments in nude mice with PSMA-expressing xenografts. Contrast Media Mol. Imaging 2015, 10, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.; Li, L.; Chea, J.; Delgado, M.K.; Crow, D.; Poku, E.; Szpikowska, B.; Bowles, N.; Channappa, D.; Colcher, D.; et al. PET imaging of (64)Cu-DOTA-scFv-anti-PSMA lipid nanoparticles (LNPs): Enhanced tumor targeting over anti-PSMA scFv or untargeted LNPs. Nucl. Med. Biol. 2017, 47, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viola-Villegas, N.T.; Sevak, K.K.; Carlin, S.D.; Doran, M.G.; Evans, H.W.; Bartlett, D.W.; Wu, A.M.; Lewis, J.S. Noninvasive Imaging of PSMA in prostate tumors with (89)Zr-Labeled huJ591 engineered antibody fragments: The faster alternatives. Mol. Pharm. 2014, 11, 3965–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit-Taskar, N.; O’Donoghue, J.A.; Ruan, S.; Lyashchenko, S.K.; Carrasquillo, J.A.; Heller, G.; Martinez, D.F.; Cheal, S.M.; Lewis, J.S.; Fleisher, M.; et al. First-in-Human Imaging with 89Zr-Df-IAB2M Anti-PSMA Minibody in Patients with Metastatic Prostate Cancer: Pharmacokinetics, Biodistribution, Dosimetry, and Lesion Uptake. J. Nucl. Med. 2016, 57, 1858–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Yu, L.; Liu, N.; Guo, Z.; Wang, G.; Zheng, J.; Wei, M.; Wang, H.; Yang, A.G.; Qin, W.; et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett. 2013, 338, 282–291. [Google Scholar] [CrossRef]
- Baum, V.; Buhler, P.; Gierschner, D.; Herchenbach, D.; Fiala, G.J.; Schamel, W.W.; Wolf, P.; Elsasser-Beile, U. Antitumor activities of PSMAxCD3 diabodies by redirected T-cell lysis of prostate cancer cells. Immunotherapy 2013, 5, 27–38. [Google Scholar] [CrossRef]
- Friedrich, M.; Raum, T.; Lutterbuese, R.; Voelkel, M.; Deegen, P.; Rau, D.; Kischel, R.; Hoffmann, P.; Brandl, C.; Schuhmacher, J.; et al. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol. Cancer Ther. 2012, 11, 2664–2673. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Hoyos, G.; Sewell, T.; Bader, R.; Bannink, J.; Chenault, R.A.; Daugherty, M.; Dasovich, M.; Fang, H.; Gottschalk, R.; Kumer, J.; et al. MOR209/ES414, a Novel Bispecific Antibody Targeting PSMA for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2155–2165. [Google Scholar] [CrossRef] [Green Version]
- Jachimowicz, R.D.; Fracasso, G.; Yazaki, P.J.; Power, B.E.; Borchmann, P.; Engert, A.; Hansen, H.P.; Reiners, K.S.; Marie, M.; von Strandmann, E.P.; et al. Induction of in vitro and in vivo NK cell cytotoxicity using high-avidity immunoligands targeting prostate-specific membrane antigen in prostate carcinoma. Mol. Cancer Ther. 2011, 10, 1036–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leconet, W.; Liu, H.; Guo, M.; Le Lamer-Dechamps, S.; Molinier, C.; Kim, S.; Vrlinic, T.; Oster, M.; Liu, F.; Navarro, V.; et al. Anti-PSMA/CD3 Bispecific Antibody Delivery and Antitumor Activity Using a Polymeric Depot Formulation. Mol. Cancer Ther. 2018, 17, 1927–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Helfand, B.T.; Carneiro, B.A.; Qin, W.; Yang, X.J.; Lee, C.; Zhang, W.; Giles, F.J.; Cristofanilli, M.; Kuzel, T.M. Efficacy Against Human Prostate Cancer by Prostate-specific Membrane Antigen-specific, Transforming Growth Factor-beta Insensitive Genetically Targeted CD8(+) T-cells Derived from Patients with Metastatic Castrate-resistant Disease. Eur. Urol. 2018, 73, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Franek, K.J.; Patterson, A.L.; Holmes, L.M.; Burgin, K.E.; Ji, J.; Yu, X.; Wagner, T.E.; Wei, Y. Targeting foreign major histocompatibility complex molecules to tumors by tumor cell specific single chain antibody (scFv). Int. J. Oncol. 2003, 23, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Schultze-Seemann, S.; Bogatyreva, L.; Hauschke, D.; Wetterauer, U.; Wolf, P. In vitro and in vivo effects of a recombinant anti-PSMA immunotoxin in combination with docetaxel against prostate cancer. Oncotarget 2016, 7, 22531–22542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noll, T.; Schultze-Seemann, S.; Kuckuck, I.; Michalska, M.; Wolf, P. Synergistic cytotoxicity of a prostate cancer-specific immunotoxin in combination with the BH3 mimetic ABT-737. Cancer Immunol. Immunother. 2018, 67, 413–422. [Google Scholar] [CrossRef]
- Baiz, D.; Hassan, S.; Choi, Y.A.; Flores, A.; Karpova, Y.; Yancey, D.; Pullikuth, A.; Sui, G.; Sadelain, M.; Debinski, W.; et al. Combination of the PI3K inhibitor ZSTK474 with a PSMA-targeted immunotoxin accelerates apoptosis and regression of prostate cancer. Neoplasia 2013, 15, 1172–1183. [Google Scholar] [CrossRef]
- Meng, P.; Dong, Q.C.; Tan, G.G.; Wen, W.H.; Wang, H.; Zhang, G.; Wang, Y.Z.; Jing, Y.M.; Wang, C.; Qin, W.J.; et al. Anti-tumor effects of a recombinant anti-prostate specific membrane antigen immunotoxin against prostate cancer cells. BMC Urol. 2017, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Hassani, M.; Hajari Taheri, F.; Sharifzadeh, Z.; Arashkia, A.; Hadjati, J.; van Weerden, W.M.; Abdoli, S.; Modarressi, M.H.; Abolhassani, M. Engineered Jurkat Cells for Targeting Prostate-Specific Membrane Antigen on Prostate Cancer Cells by Nanobody-Based Chimeric Antigen Receptor. Iran. Biomed. J. 2020, 24, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Santoro, S.P.; Kim, S.; Motz, G.T.; Alatzoglou, D.; Li, C.; Irving, M.; Powell, D.J., Jr.; Coukos, G. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol. Res. 2015, 3, 68–84. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Avitabile, E.; Gatta, V.; Malatesta, P.; Petrovic, B.; Campadelli-Fiume, G. HSV as A Platform for the Generation of Retargeted, Armed, and Reporter-Expressing Oncolytic Viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Hasegawa, K.; Russell, S.J.; Sadelain, M.; Peng, K.W. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate 2009, 69, 1128–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakova, Z.; Foss, C.A.; Copeland, B.T.; Morath, V.; Baranova, P.; Havlinova, B.; Skerra, A.; Pomper, M.G.; Barinka, C. Novel Monoclonal Antibodies Recognizing Human Prostate-Specific Membrane Antigen (PSMA) as Research and Theranostic Tools. Prostate 2017, 77, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Kumar, V.; Lisok, A.; Plyku, D.; Novakova, Z.; Brummet, M.; Wharram, B.; Barinka, C.; Hobbs, R.; Pomper, M.G. Evaluation of (111)In-DOTA-5D3, a Surrogate SPECT Imaging Agent for Radioimmunotherapy of Prostate-Specific Membrane Antigen. J. Nucl. Med. 2019, 60, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.T.; Guo, X.; Barinka, C.; Lupold, S.E.; Pomper, M.G.; Gabrielson, K.; Raman, V.; Artemov, D.; Hapuarachchige, S. Development of 5D3-DM1: A Novel Anti-Prostate-Specific Membrane Antigen Antibody-Drug Conjugate for PSMA-Positive Prostate Cancer Therapy. Mol. Pharm. 2020, 17, 3392–3402. [Google Scholar] [CrossRef]
- Hapuarachchige, S.; Huang, C.T.; Donnelly, M.C.; Barinka, C.; Lupold, S.E.; Pomper, M.G.; Artemov, D. Cellular Delivery of Bioorthogonal Pretargeting Therapeutics in PSMA-Positive Prostate Cancer. Mol. Pharm. 2020, 17, 98–108. [Google Scholar] [CrossRef]
- Schiweck, W.; Skerra, A. Fermenter production of an artificial fab fragment, rationally designed for the antigen cystatin, and its optimized crystallization through constant domain shuffling. Proteins 1995, 23, 561–565. [Google Scholar] [CrossRef]
- Skerra, A. Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 1994, 151, 131–135. [Google Scholar] [CrossRef]
- Schlapschy, M.; Fiedler, M.; Skerra, A. Purification and Characterization of His-Tagged Antibody Fragments in Antibody Engineering, 2nd ed.; Springer: Berlin, Germany, 2010; pp. 279–291. [Google Scholar] [CrossRef]
- Chilumuri, A.; Markiv, A.; Milton, N.G. Immunocytochemical staining of endogenous nuclear proteins with the HIS-1 anti-poly-histidine monoclonal antibody: A potential source of error in His-tagged protein detection. Acta Histochem. 2014, 116, 1022–1028. [Google Scholar] [CrossRef]
- Vermeer, A.W.; Bremer, M.G.; Norde, W. Structural changes of IgG induced by heat treatment and by adsorption onto a hydrophobic Teflon surface studied by circular dichroism spectroscopy. Biochi. Biophys. Acta 1998, 1425, 1–12. [Google Scholar] [CrossRef]
- Gupta, S.K.; Shukla, P. Microbial platform technology for recombinant antibody fragment production: A review. Crit. Rev. Microbiol. 2017, 43, 31–42. [Google Scholar] [CrossRef] [PubMed]
- De Jongh, W.A.; Salgueiro, S.; Dyring, C. The use of Drosophila S2 cells in R&D and bioprocessing. Pharm. Bioprocess. 2013, 1, 197–213. [Google Scholar]
- Tykvart, J.; Sacha, P.; Barinka, C.; Knedlik, T.; Starkova, J.; Lubkowski, J.; Konvalinka, J. Efficient and versatile one-step affinity purification of in vivo biotinylated proteins: Expression, characterization and structure analysis of recombinant human glutamate carboxypeptidase II. Protein Expr. Purif. 2012, 82, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventini-Monteiro, D.; Dubois, S.; Astray, R.M.; Castillo, J.; Pereira, C.A. Insect cell entrapment, growth and recovering using a single-use fixed-bed bioreactor. Scaling up and recombinant protein production. J. Biotechnol. 2015, 216, 110–115. [Google Scholar] [CrossRef]
- Daniels, R.W.; Rossano, A.J.; Macleod, G.T.; Ganetzky, B. Expression of multiple transgenes from a single construct using viral 2A peptides in Drosophila. PLoS ONE 2014, 9, e100637. [Google Scholar] [CrossRef]
- Mori, K.; Hamada, H.; Ogawa, T.; Ohmuro-Matsuyama, Y.; Katsuda, T.; Yamaji, H. Efficient production of antibody Fab fragment by transient gene expression in insect cells. J. Biosci. Bioeng. 2017, 124, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [Green Version]
- Viti, F.; Tarli, L.; Giovannoni, L.; Zardi, L.; Neri, D. Increased binding affinity and valence of recombinant antibody fragments lead to improved targeting of tumoral angiogenesis. Cancer Res. 1999, 59, 347–352. [Google Scholar]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [Green Version]
- Whitlow, M.; Bell, B.A.; Feng, S.L.; Filpula, D.; Hardman, K.D.; Hubert, S.L.; Rollence, M.L.; Wood, J.F.; Schott, M.E.; Milenic, D.E.; et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993, 6, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Olafsen, T.; Kenanova, V.E.; Wu, A.M. Generation of Single-Chain Fv Fragments and Multivalent Derivatives scFv-Fc and scFv-CH3 (Minibodies) in Antibody Engineering, 2nd ed.; Springer: Berlin, Germany, 2010; pp. 69–84. [Google Scholar] [CrossRef]
- Kellmann, S.J.; Dubel, S.; Thie, H. A strategy to identify linker-based modules for the allosteric regulation of antibody-antigen binding affinities of different scFvs. MAbs 2017, 9, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Muller, D. scFv by Two-Step Cloning in Antibody Engineering, 2nd ed.; Springer: Berlin, Germany, 2010; pp. 55–59. [Google Scholar] [CrossRef]
- Martoglio, B.; Dobberstein, B. Signal sequences: More than just greasy peptides. Trends Cell Biol. 1998, 8, 410–415. [Google Scholar] [CrossRef]
- El-Sayed, A.; Bernhard, W.; Barreto, K.; Gonzalez, C.; Hill, W.; Pastushok, L.; Fonge, H.; Geyer, C.R. Evaluation of antibody fragment properties for near-infrared fluorescence imaging of HER3-positive cancer xenografts. Theranostics 2018, 8, 4856–4869. [Google Scholar] [CrossRef] [PubMed]
- Pavlinkova, G.; Beresford, G.W.; Booth, B.J.; Batra, S.K.; Colcher, D. Pharmacokinetics and biodistribution of engineered single-chain antibody constructs of MAb CC49 in colon carcinoma xenografts. J. Nucl. Med. 1999, 40, 1536–1546. [Google Scholar]
- Adams, G.P.; Schier, R.; Marshall, K.; Wolf, E.J.; McCall, A.M.; Marks, J.D.; Weiner, L.M. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998, 58, 485–490. [Google Scholar]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar]
- Zhou, Y.; Goenaga, A.L.; Harms, B.D.; Zou, H.; Lou, J.; Conrad, F.; Adams, G.P.; Schoeberl, B.; Nielsen, U.B.; Marks, J.D. Impact of intrinsic affinity on functional binding and biological activity of EGFR antibodies. Mol. Cancer Ther. 2012, 11, 1467–1476. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Wu, J.; Han, Y.; Wei, M.; Han, S.; Lin, R.; Sun, Z.; Yang, F.; Jiao, D.; Xie, P.; et al. A novel anti-PSMA human scFv has the potential to be used as a diagnostic tool in prostate cancer. Oncotarget 2016, 7, 59471–59481. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, J.; RajabiBazl, M.; Ebrahimizadeh, W.; Dehbidi, G.R.; Hosseini, H. Selection of Single Chain Antibody Fragments for Targeting Prostate Specific Membrane Antigen: A Comparison Between Cell-based and Antigen-based Approach. Protein Pept. Lett. 2016, 23, 336–342. [Google Scholar] [CrossRef]
- Frigerio, B.; Fracasso, G.; Luison, E.; Cingarlini, S.; Mortarino, M.; Coliva, A.; Seregni, E.; Bombardieri, E.; Zuccolotto, G.; Rosato, A.; et al. A single-chain fragment against prostate specific membrane antigen as a tool to build theranostic reagents for prostate cancer. Eur. J. Cancer 2013, 49, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.A.; Diaz, I.L.; Anderson, K.A.; Batt, C.A. Design, production, and characterization of a single-chain variable fragment (ScFv) derived from the prostate specific membrane antigen (PSMA) monoclonal antibody J591. Protein Expr. Purif. 2013, 89, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Kampmeier, F.; Williams, J.D.; Maher, J.; Mullen, G.E.; Blower, P.J. Design and preclinical evaluation of a 99mTc-labelled diabody of mAb J591 for SPECT imaging of prostate-specific membrane antigen (PSMA). EJNMMI Res. 2014, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.A.; Comeau, R.D.; Jones, P.L.; Liberatore, F.A.; Neacy, W.P.; Sands, H.; Gallagher, B.M. Pharmacokinetics of the monoclonal antibody B72.3 and its fragments labeled with either 125I or 111In. Cancer Res. 1987, 47, 1149–1154. [Google Scholar] [PubMed]
- Fortmuller, K.; Alt, K.; Gierschner, D.; Wolf, P.; Baum, V.; Freudenberg, N.; Wetterauer, U.; Elsasser-Beile, U.; Buhler, P. Effective targeting of prostate cancer by lymphocytes redirected by a PSMA x CD3 bispecific single-chain diabody. Prostate 2011, 71, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Werner, W.E.; Wu, S.; Mulkerrin, M. The removal of pyroglutamic acid from monoclonal antibodies without denaturation of the protein chains. Anal. Biochem. 2005, 342, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef]
- Strohalm, M.; Hassman, M.; Kosata, B.; Kodicek, M. mMass data miner: An open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012, 503, 157–188. [Google Scholar] [CrossRef]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Barinka, C.; Ptacek, J.; Richter, A.; Novakova, Z.; Morath, V.; Skerra, A. Selection and characterization of Anticalins targeting human prostate-specific membrane antigen (PSMA). Protein Eng. Des. Sel. 2016, 29, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.R.; Foss, C.A.; Castanares, M.; Mease, R.C.; Byun, Y.; Fox, J.J.; Hilton, J.; Lupold, S.E.; Kozikowski, A.P.; Pomper, M.G. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA). J. Med. chem. 2008, 51, 4504–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| appKD (nM) | ||
|---|---|---|
| 5D3 Variants | ELISA | Flow Cytometry |
| Fab | 1.0 ± 0.7 | 1.5 ± 0.2 |
| scFv HL | 2.3 ± 1.7 | 1.5 ± 0.1 |
| scFv LH | 2.4 ± 0.1 | 1.3 ± 0.5 |
| mAb | 0.23 ± 0.1 | 0.85 ± 0.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novakova, Z.; Belousova, N.; Foss, C.A.; Havlinova, B.; Gresova, M.; Das, G.; Lisok, A.; Prada, A.; Barinkova, M.; Hubalek, M.; et al. Engineered Fragments of the PSMA-Specific 5D3 Antibody and Their Functional Characterization. Int. J. Mol. Sci. 2020, 21, 6672. https://doi.org/10.3390/ijms21186672
Novakova Z, Belousova N, Foss CA, Havlinova B, Gresova M, Das G, Lisok A, Prada A, Barinkova M, Hubalek M, et al. Engineered Fragments of the PSMA-Specific 5D3 Antibody and Their Functional Characterization. International Journal of Molecular Sciences. 2020; 21(18):6672. https://doi.org/10.3390/ijms21186672
Chicago/Turabian StyleNovakova, Zora, Nikola Belousova, Catherine A. Foss, Barbora Havlinova, Marketa Gresova, Gargi Das, Ala Lisok, Adam Prada, Marketa Barinkova, Martin Hubalek, and et al. 2020. "Engineered Fragments of the PSMA-Specific 5D3 Antibody and Their Functional Characterization" International Journal of Molecular Sciences 21, no. 18: 6672. https://doi.org/10.3390/ijms21186672