Oxidative Photocyclization of Aromatic Schiff Bases in Synthesis of Phenanthridines and Other Aza-PAHs †

 , , and
, , and
Abstract
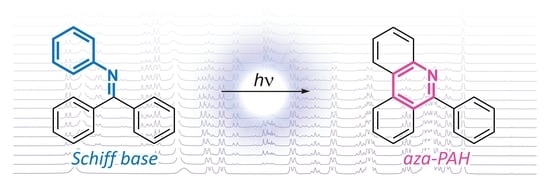
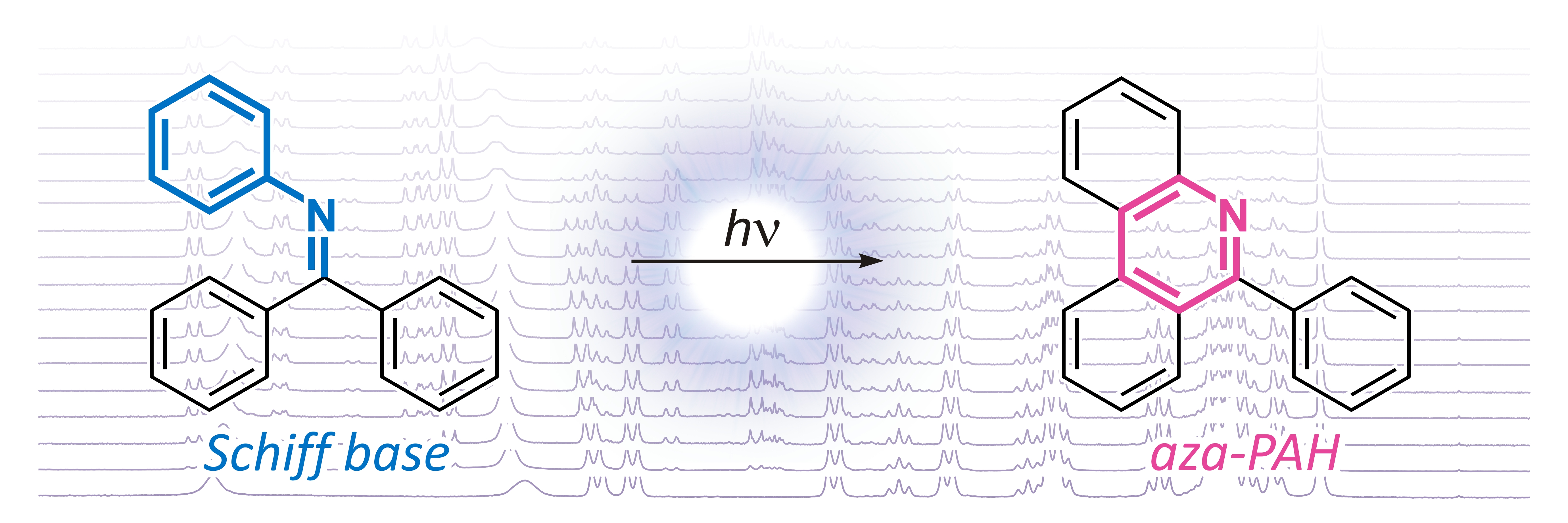
:
1. Introduction
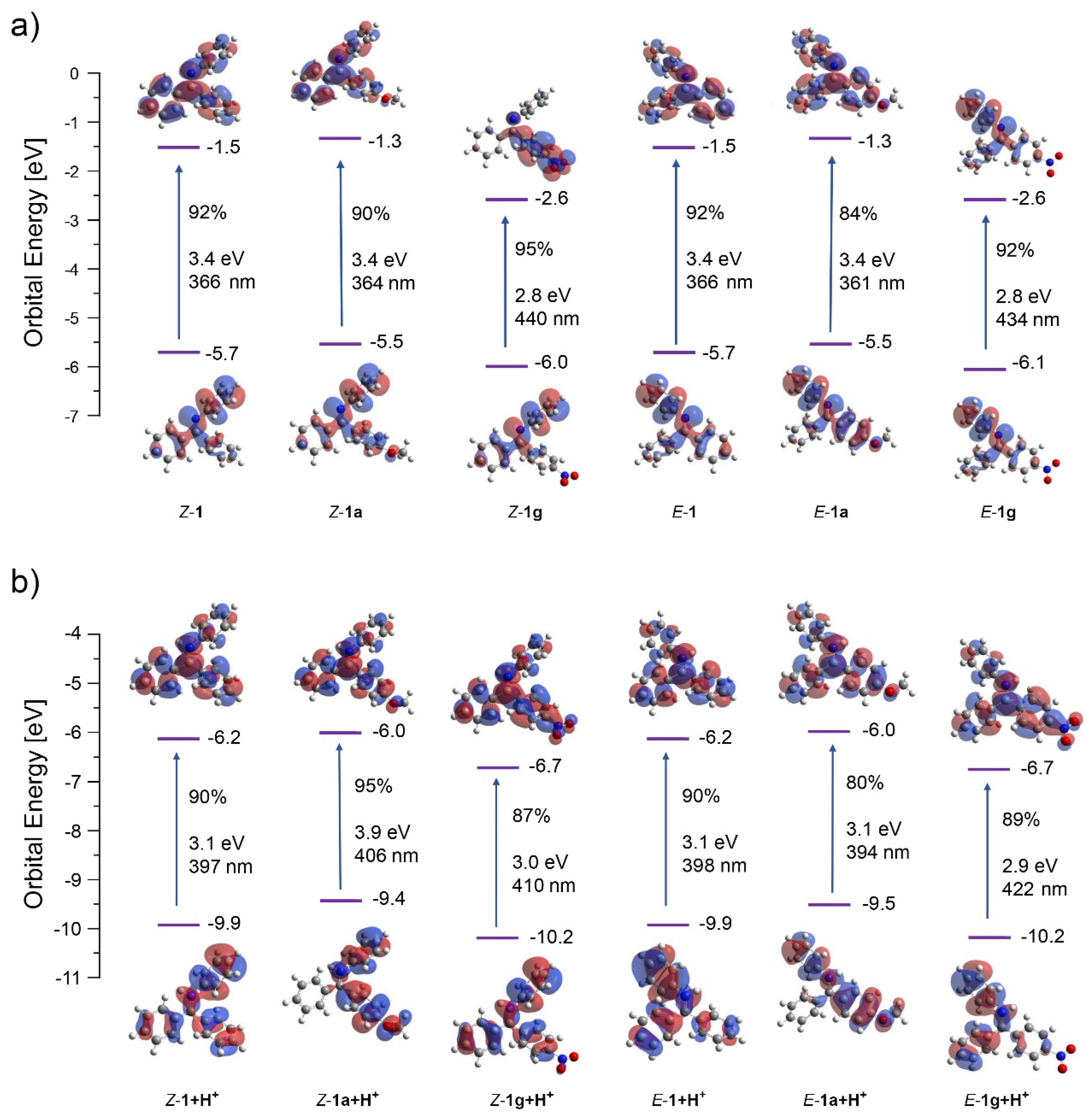
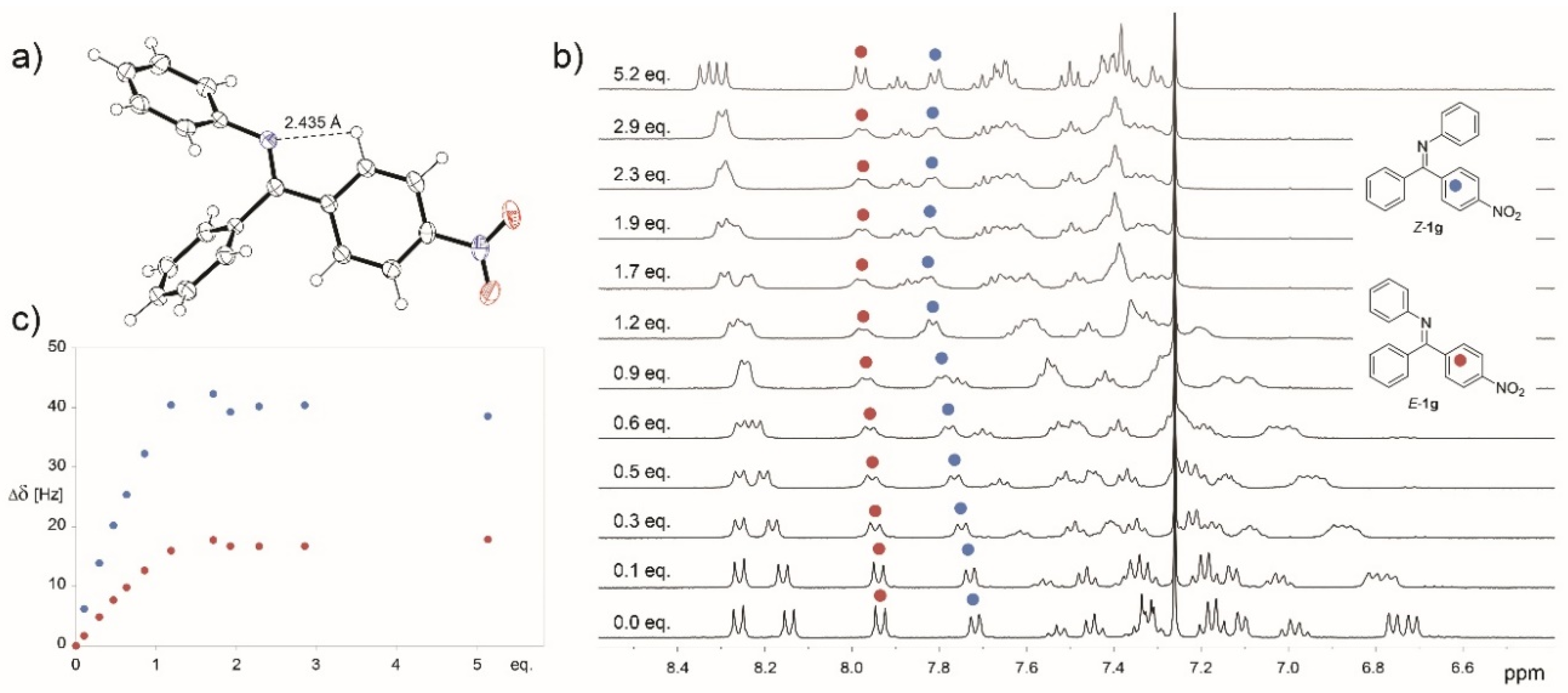
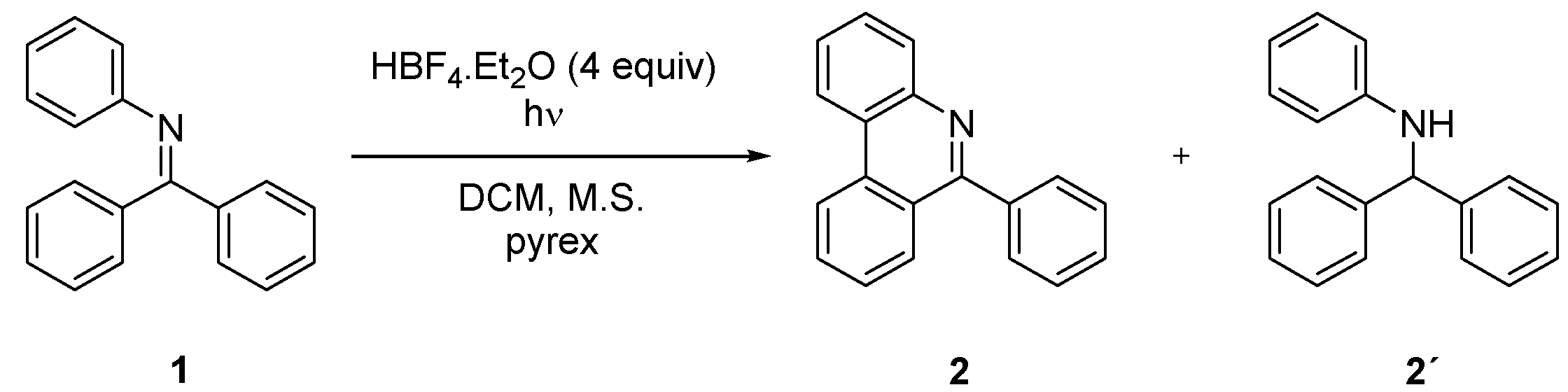
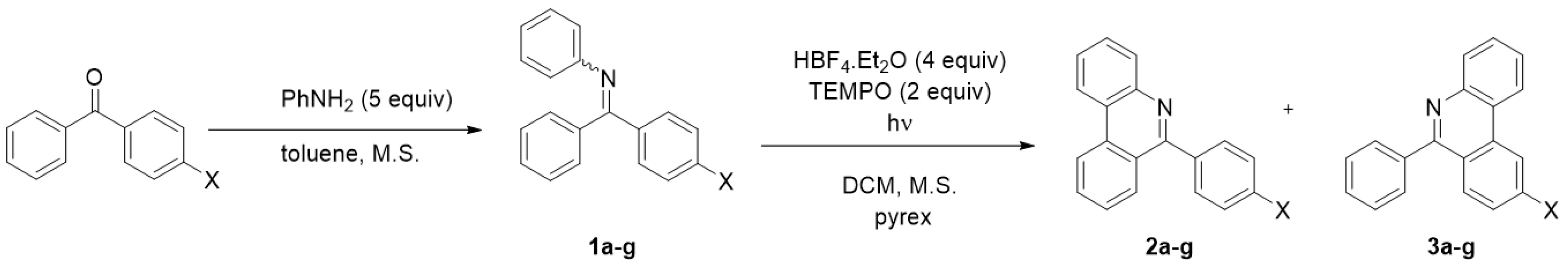
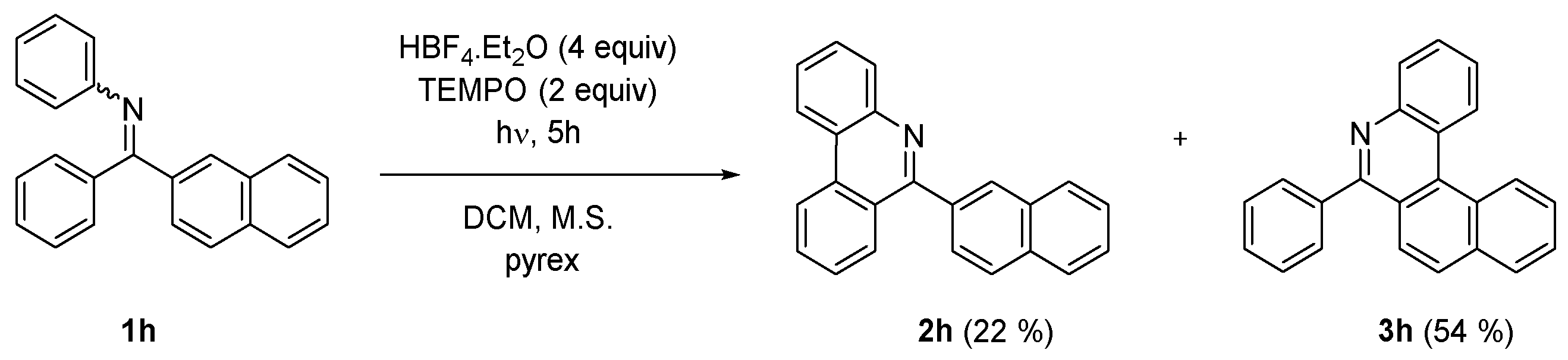
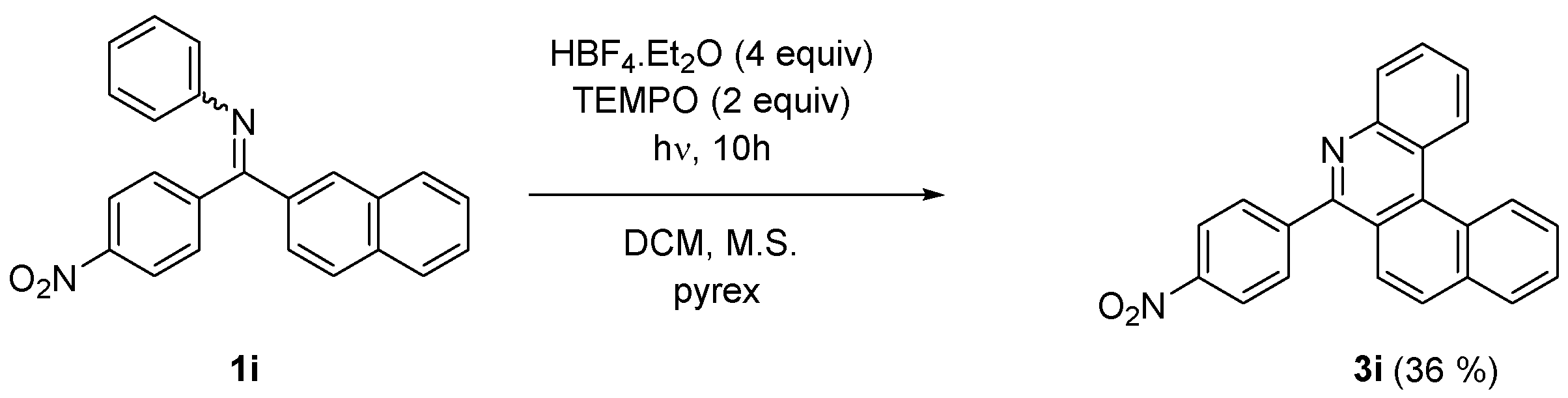
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APCI | Atmospheric pressure chemical ionization |
| dba | Dibenzylidenaceton |
| DCM | Dichloromethane |
| DMSO | Dimethyl sulfoxide |
| ESI | Electrospray ionization |
| GC-MS | Gas chromatography–mass spectrometry |
| HOMO | Highest occupied molecular orbital |
| IR | Infrared |
| LUMO | Lowest unoccupied molecular orbital |
| M.S. | Molecular sieves |
| NMP | N-Methyl-2-pyrrolidone |
| NMR | Nuclear magnetic resonance |
| NOESY | Nuclear Overhauser effect spectroscopy |
| ORTEP | Oak ridge thermal ellipsoid plot |
| PAH | Polycyclic aromatic hydrocarbon |
| PTFE | Polytetrafluoroethylene |
| TD-DFT | Time-dependent density functional theory |
| TEMPO | 2,2,6,6-Tetramethyl-1-piperidinyloxy |
| THF | Tetrahydrofuran |
| TLC | Thin-layer chromatography |
| TMS | Tetramethylsilane |
| TOF | Time of flight |
| UV | Ultraviolet |
| XPhos | 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl |
References
- Nakanishi, T.; Masuda, A.; Suwa, M.; Akiyama, Y.; Hoshino-Abe, N.; Suzuki, M. Synthesis of derivatives of NK109, 7-OH benzo[c]phenanthridine alkaloid, and evaluation of their cytotoxicities and reduction-resistant properties. Bioorg. Med. Chem. Lett. 2000, 10, 2321–2323. [Google Scholar] [CrossRef]
- Phillips, S.D.; Castle, R.N. A Review of the chemistry of the antitumor Benzo[c]phenanthridine Alkaloids Nitidine and Fagaronine an of the related antitumor alkaloid coralyne. J. Heterocycl. Chem. 1981, 18, 223–232. [Google Scholar] [CrossRef]
- Bernardo, P.H.; Wan, K.F.; Sivaraman, T.; Xu, J.; Moore, F.K.; Hung, A.W.; Mok, H.Y.K.; Yu, V.C.; Chai, C.L.L. Structure-activity relationship studies of phenanthridine-based Bcl-XL inhibitors. J. Med. Chem. 2008, 51, 6699–6710. [Google Scholar] [CrossRef]
- Nakanishi, T.; Suzuki, M.; Saimoto, A.; Kabasawa, T. Structural considerations of NK109, an antitumor benzo[c]phenanthridine alkaloid. J. Nat. Prod. 1999, 62, 864–867. [Google Scholar] [CrossRef]
- Abdel-Halim, O.B.; Morikawa, T.; Ando, S.; Matsuda, H.; Yoshikawa, M. New crinine-type alkaloids with inhibitory effect on induction of inducible nitric oxide synthase from Crinum yemense. J. Nat. Prod. 2004, 67, 1119–1124. [Google Scholar] [CrossRef]
- Stevens, N.; O’Connor, N.; Vishwasrao, H.; Samaroo, D.; Kandel, E.R.; Akins, D.L.; Drain, C.M.; Turro, N.J. Two Color RNA Intercalating Probe for Cell Imaging Applications. J. Am. Chem. Soc. 2008, 130, 7182–7183. [Google Scholar] [CrossRef] [PubMed]
- Tumir, L.M.; Stojković, M.R.; Piantanida, I. Come-back of phenanthridine and phenanthridinium derivatives in the 21st century. Beilstein J. Org. Chem. 2014, 10, 2930–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Cheng, Y.; Wang, R.; Zheng, M.; Zhang, Y.; Yu, S. Synthesis of 6-alkylated phenanthridine derivatives using photoredox neutral somophilic isocyanide insertion. Angew. Chem. Int. Ed. 2013, 52, 13289–13292. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, D.; Huang, W.; Sun, H.; Wang, L.; Ren, M.; Wang, B.; Ma, Y. Metal-free photocatalyzed cross coupling of aryl (heteroaryl) bromides with isonitriles. Tetrahedron 2017, 73, 7094–7099. [Google Scholar] [CrossRef]
- Lysén, M.; Kristensen, J.L.; Vedsø, P.; Begtrup, M. Convergent synthesis of 6-substituted phenanthridines via anionic ring closure. Org. Lett. 2002, 4, 257–259. [Google Scholar] [CrossRef]
- Bowman, W.R.; Lyon, J.E.; Pritchard, G.J. Palladium and radical routes to phenanthridines. Arkivoc 2012, 7, 210–227. [Google Scholar] [CrossRef]
- Evoniuk, C.J.; Gomes, G.D.P.; Ly, M.; White, F.D.; Alabugin, I.V. Coupling Radical Homoallylic Expansions with C-C Fragmentations for the Synthesis of Heteroaromatics: Quinolines from Reactions of o-Alkenylarylisonitriles with Aryl, Alkyl, and Perfluoroalkyl Radicals. J. Org. Chem. 2017, 82, 4265–4278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dong, X.; Xiao, T.; Zhou, L. PhI(OAc) 2 -Mediated Synthesis of 6-(Trifluoromethyl)phenanthridines by Oxidative Cyclization of 2-Isocyanobiphenyls with CF3SiMe3 under Metal-Free Conditions. Org. Lett. 2013, 15, 4846–4849. [Google Scholar] [CrossRef] [PubMed]
- Tobisu, M.; Koh, K.; Furukawa, T.; Chatani, N. Modular synthesis of phenanthridine derivatives by oxidative cyclization of 2-isocyanobiphenyls with organoboron reagents. Angew. Chem. Int. Ed. 2012, 51, 11363–11366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Mück-Lichtenfeld, C.; Daniliuc, C.G.; Studer, A. 6-Trifluoromethyl-phenanthridines through radical trifluoromethylation of isonitriles. Angew. Chem. Int. Ed. 2013, 52, 10792–10795. [Google Scholar] [CrossRef] [PubMed]
- Leifert, D.; Daniliuc, C.G.; Studer, A. 6-Aroylated phenanthridines via base promoted homolytic aromatic substitution (BHAS). Org. Lett. 2013, 15, 6286–6289. [Google Scholar] [CrossRef]
- Pan, C.; Zhang, H.; Han, J.; Cheng, Y.; Zhu, C. Metal-free radical oxidative decarboxylation/cyclization of acyl peroxides and 2-isocyanobiphenyls. Chem. Commun. 2015, 51, 3786–3788. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, C.; Dong, X.; Qu, J. Synthesis of 6-Trichloromethylphenanthridines by Transition Metal-Free Radical Cyclization of 2-Isocyanobiphenyls. J. Org. Chem. 2016, 81, 5202–5208. [Google Scholar] [CrossRef]
- Youn, S.W.; Bihn, J.H. Trifluoroacetic acid-mediated facile construction of 6-substituted phenanthridines. Tetrahedron Lett. 2009, 50, 4598–4601. [Google Scholar] [CrossRef]
- Zheng, Y.-H.; Lu, H.-Y.; Li, M.; Chen, C.-F. Synthesis, Structures, and Optical Properties of Aza[4]helicenes. Eur. J. Org. Chem. 2013, 2013, 3059–3066. [Google Scholar] [CrossRef]
- Tang, C.; Yuan, Y.; Jiao, N. Metal-free nitrogenation of 2-acetylbiphenyls: Expeditious synthesis of phenanthridines. Org. Lett. 2015, 17, 2206–2209. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.L.; Wu, Y.; Wong, S.M.; Kwong, F.Y.; Mao, F. Intramolecular Direct C–H Bond Arylation from Aryl Chlorides: A Transition-Metal-Free Approach for Facile Access of Phenanthridines. Org. Lett. 2012, 14, 5306–5309. [Google Scholar] [CrossRef]
- Peng, J.; Chen, T.; Chen, C.; Li, B. Palladium-Catalyzed Intramolecular C–H Activation/C–C Bond Formation: A Straightforward Synthesis of Phenanthridines. J. Org. Chem. 2011, 76, 9507–9513. [Google Scholar] [CrossRef] [PubMed]
- Evoniuk, C.J.; Gomes, G.D.P.; Hill, S.P.; Fujita, S.; Hanson, K.; Alabugin, I.V. Coupling N–H Deprotonation, C–H Activation, and Oxidation: Metal-Free C(sp 3)–H Aminations with Unprotected Anilines. J. Am. Chem. Soc. 2017, 139, 16210–16221. [Google Scholar] [CrossRef] [PubMed]
- Evoniuk, C.J.; Hill, S.P.; Hanson, K.; Alabugin, I.V. Double C-H amination by consecutive SET oxidations. Chem. Commun. 2016, 52, 7138–7141. [Google Scholar] [CrossRef] [PubMed]
- Mallory, F.B.; Mallory, C.W. Photocyclization of Stilbenes and Related Molecules. Org. React. 1984, 30, 1–456. [Google Scholar] [CrossRef]
- Hugelshofer, P.; Kalvoda, J.; Schaffner, K. Photochemische Reaktionen. 8. Mitteilung. Lichtkatalysierte Cyclodehydrierung von 1,2-Diaryläthylenen und Azobenzol. Helv. Chim. Acta 1960, 43, 1322–1332. [Google Scholar] [CrossRef]
- Mallory, F.B.; Wood, C.S. The Photocyclization of Anils to Phenanthridines. Tetrahedron Lett. 1965, 6, 2643–2648. [Google Scholar] [CrossRef]
- Pratt, A.C. The photochemistry of imines. Chem. Soc. Rev. 1977, 6, 63–81. [Google Scholar] [CrossRef]
- Badger, G.M.; Joshua, C.P.; Lewis, G.E. Photocatalysed cyclization of benzalaniline. Tetrahedron Lett. 1964, 5, 3711–3713. [Google Scholar] [CrossRef]
- Perkampus, H.-H.; Behjati, B. Darstellung einiger diazaphenanthrene durch photocyclisierung der benzylidenaminopyridine und pyridinalaniline. J. Heterocycl. Chem. 1974, 11, 511–514. [Google Scholar] [CrossRef]
- Scholz, M.; Dietz, F.; Mühlstädt, M. Chemie angeregter zustände. V. mitt.: Photocyclisierung von benzalanthronen. Tetrahedron Lett. 1970, 11, 2835–2838. [Google Scholar] [CrossRef]
- Thompson, C.M.; Docter, S. Lewis acid promoted photocyclization of arylimines. Studies directed towards the synthesis of pentacyclic natural products. Tetrahedron Lett. 1988, 29, 5213–5216. [Google Scholar] [CrossRef]
- Yang, Y.; Da Costa, R.C.; Smilgies, D.M.; Campbell, A.J.; Fuchter, M.J. Induction of circularly polarized electroluminescence from an achiral light-emitting polymer via a chiral small-molecule dopant. Adv. Mater. 2013, 25, 2624–2628. [Google Scholar] [CrossRef] [Green Version]
- Wallabregue, A.; Sherin, P.; Guin, J.; Besnard, C.; Vauthey, E.; Lacour, J. Modular Synthesis of pH-Sensitive Fluorescent Diaza[4]helicenes. Eur. J. Org. Chem. 2014, 2014, 6431–6438. [Google Scholar] [CrossRef]
- Saleh, N.; Moore, B.; Srebro, M.; Vanthuyne, N.; Toupet, L.; Williams, J.A.G.; Roussel, C.; Deol, K.K.; Muller, G.; Autschbach, J.; et al. Acid/Base-Triggered Switching of Circularly Polarized Luminescence and Electronic Circular Dichroism in Organic and Organometallic Helicenes. Chem. A Eur. J. 2015, 21, 1673–1681. [Google Scholar] [CrossRef]
- Matsushima, T.; Kobayashi, S.; Watanabe, S. Air-Driven Potassium Iodide-Mediated Oxidative Photocyclization of Stilbene Derivatives. J. Org. Chem. 2016, 81, 7799–7806. [Google Scholar] [CrossRef]
- Žádný, J.; Velíšek, P.; Jakubec, M.; Sýkora, J.; Církva, V.; Storch, J. Exploration of 9-bromo[7]helicene reactivity. Tetrahedron 2013, 69, 6213–6218. [Google Scholar] [CrossRef]
- Amal Joseph, P.J.; Priyadarshini, S.; Lakshmi Kantam, M.; Maheswaran, H. Copper catalyzed ipso-nitration of iodoarenes, bromoarenes and heterocyclic haloarenes under ligand-free conditions. Tetrahedron Lett. 2012, 53, 1511–1513. [Google Scholar] [CrossRef]
- Huang, X.; Anderson, K.W.; Zim, D.; Jiang, L.; Klapars, A.; Buchwald, S.L. Expanding Pd-catalyzed C-N bond-forming processes: The first amidation of aryl sulfonates, aqueous amination, and complementarity with Cu-catalyzed reactions. J. Am. Chem. Soc. 2003, 125, 6653–6655. [Google Scholar] [CrossRef]
- Církva, V.; Jakubík, P.; Strašák, T.; Hrbáč, J.; Sýkora, J.; Císařová, I.; Vacek, J.; Žádný, J.; Storch, J. Preparation and Physicochemical Properties of [6]Helicenes Fluorinated at Terminal Rings. J. Org. Chem. 2019, 84, 1980–1993. [Google Scholar] [CrossRef] [PubMed]
- Talele, H.R.; Chaudhary, A.R.; Patel, P.R.; Bedekar, A.V. Expeditious synthesis of helicenes using an improved protocol of photocyclodehydrogenation of stilbenes. Arkivoc 2011, 9, 15–37. [Google Scholar] [CrossRef]
- Laarhoven, W.H.; Cuppen, T.H.J.H.M.; Nivard, R.J.F. Photodehydrocyclizations in stilbene-like compounds-III. Effect of steric factors. Tetrahedron 1970, 26, 4865–4881. [Google Scholar] [CrossRef]
- Ito, N.; Hirose, T.; Matsuda, K. Facile photochemical synthesis of 5,10-disubstituted [5]helicenes by removing molecular orbital degeneracy. Org. Lett. 2014, 16, 2502–2505. [Google Scholar] [CrossRef]
- Lefebvre, Q.; Jentsch, M.; Rueping, M. Continuous flow photocyclization of stilbenes-scalable synthesis of functionalized phenanthrenes and helicenes. Beilstein J. Org. Chem. 2013, 9, 1883–1890. [Google Scholar] [CrossRef] [Green Version]
- Jakubec, M.; Beránek, T.; Jakubík, P.; Sýkora, J.; Žádný, J.; Církva, V.; Storch, J. 2-Bromo[6]helicene as a Key Intermediate for [6]Helicene Functionalization. J. Org. Chem. 2018, 83, 3607–3616. [Google Scholar] [CrossRef]
- Hacker, A.S.; Pavano, M.; Wood, J.E.; Immoos, C.E.; Hashimoto, H.; Genis, S.P.; Frantz, D.K. Synthesis and Electronic Properties of Fluoreno[2,1-a]fluorenedione and Fluoreno[1,2-a]fluorenedione. J. Org. Chem. 2018, 83, 510–515. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
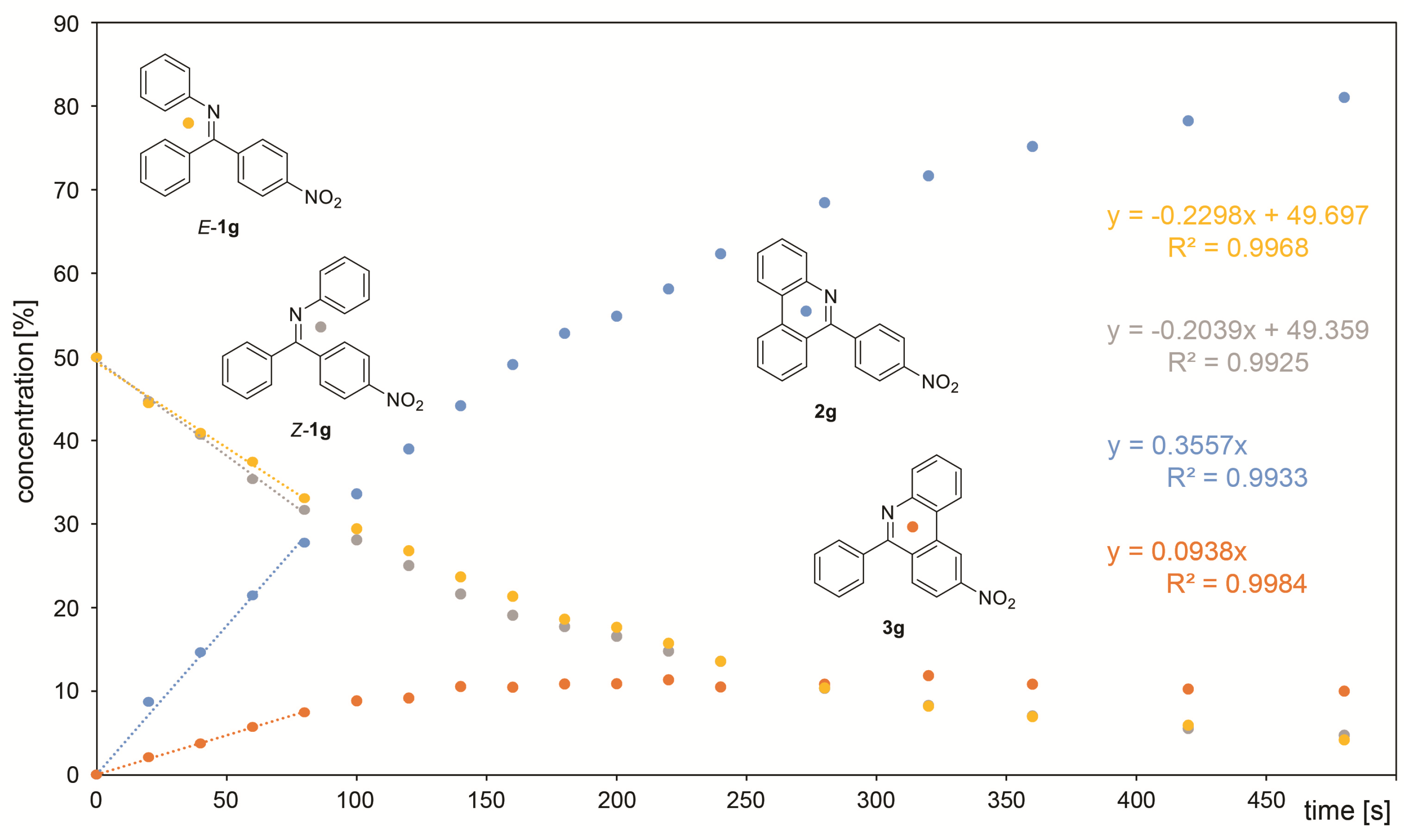
| Starting Material | X= | Reaction Time [h] | Ratio of Isomers a | Ratio of Products b 2a-g:3a-g [%] | Sum of Isolated Yields [%] |
|---|---|---|---|---|---|
| 1a | OMe | 14 | 63:37 | 86:14 | 42 c |
| 1b | Me | 8 | 52:48 | 63:37 | 80 |
| 1 | H | 4 | - | - | 71 |
| 1c | F | 4 | 61:39 | 83:17 | 72 |
| 1d | Cl | 4 | 63:37 | 73:27 | 67 |
| 1e | Br | 5 | 56:44 | 74:26 | 70 |
| 1f | CF3 | 5 | 51:49 | 78:22 | 64 |
| 1g | NO2 | 4 | 52:48 | 90:10 | 58 c |
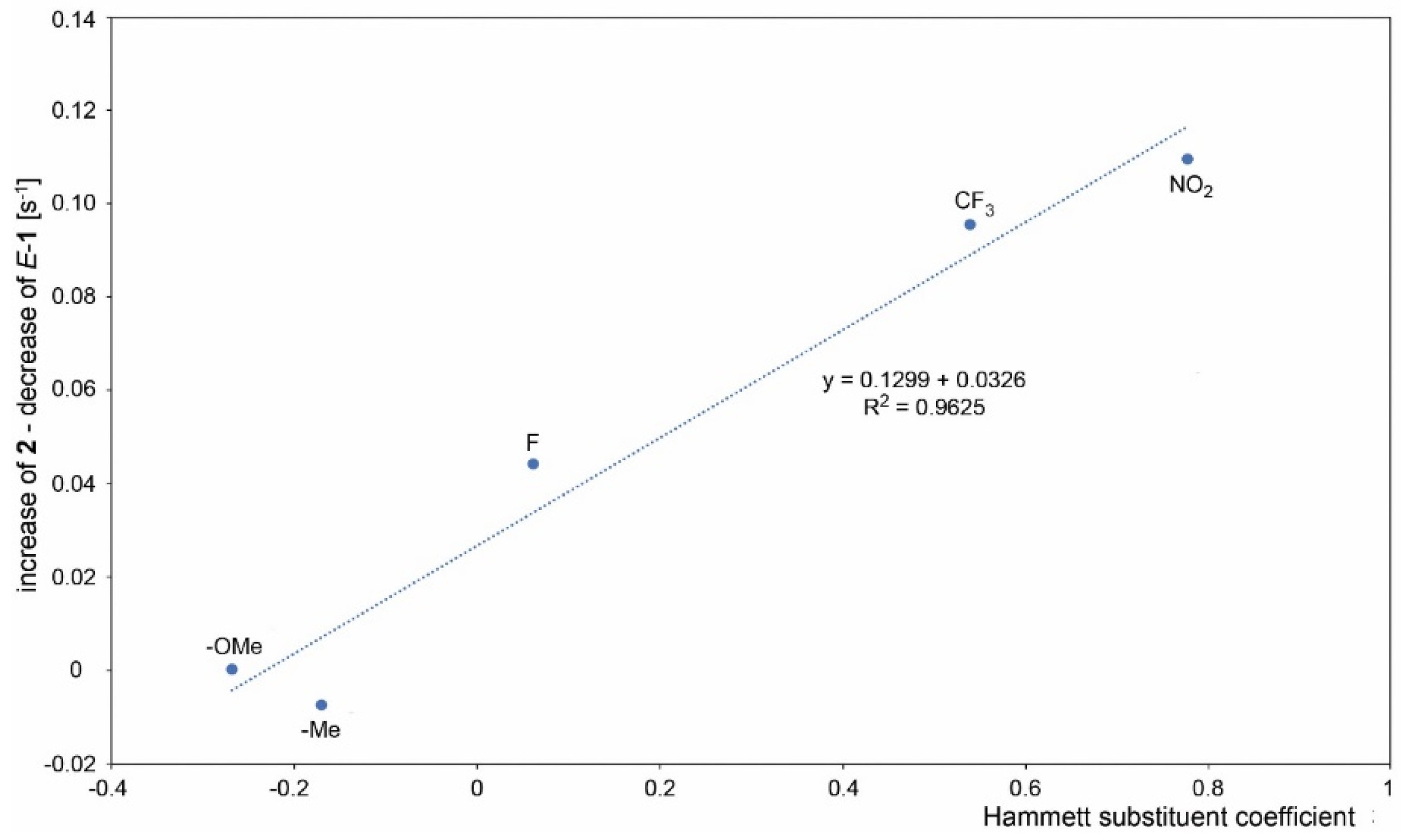
| Substituent | Hammett Substituent Constant | Rates | ||||||
|---|---|---|---|---|---|---|---|---|
| Decrease | Increase | Difference | ||||||
| Σ 1 | E-1 | Z-1 | 2 | 3 | 2—E-1 | Z-1—3 | ||
| 1a (OMe) | −0.268 | 0.039 | 0.029 | 0.010 | 0.028 | 0.003 | −0.001 | 0.007 |
| 1b (Me) | −0.170 | 0.117 | 0.077 | 0.040 | 0.072 | 0.044 | −0.006 | −0.004 |
| 1 (H) | 0 | 0.122 | - | - | 0.122 | - | - | - |
| 1c (F) | 0.062 | 0.216 | 0.111 | 0.105 | 0.159 | 0.079 | 0.048 | 0.026 |
| 1d (Cl) | 0.227 | 0.370 | - | - | 0.268 | 0.102 | - | - |
| 1e (Br) | 0.232 | 0.516 | - | - | 0.380 | 0.136 | - | - |
| 1f (CF3) | 0.540 | 0.534 | 0.307 | 0.227 | 0.417 | 0.120 | 0.110 | 0.107 |
| 1g (NO2) | 0.778 | 0.434 | 0.230 | 0.204 | 0.356 | 0.094 | 0.126 | 0.110 |
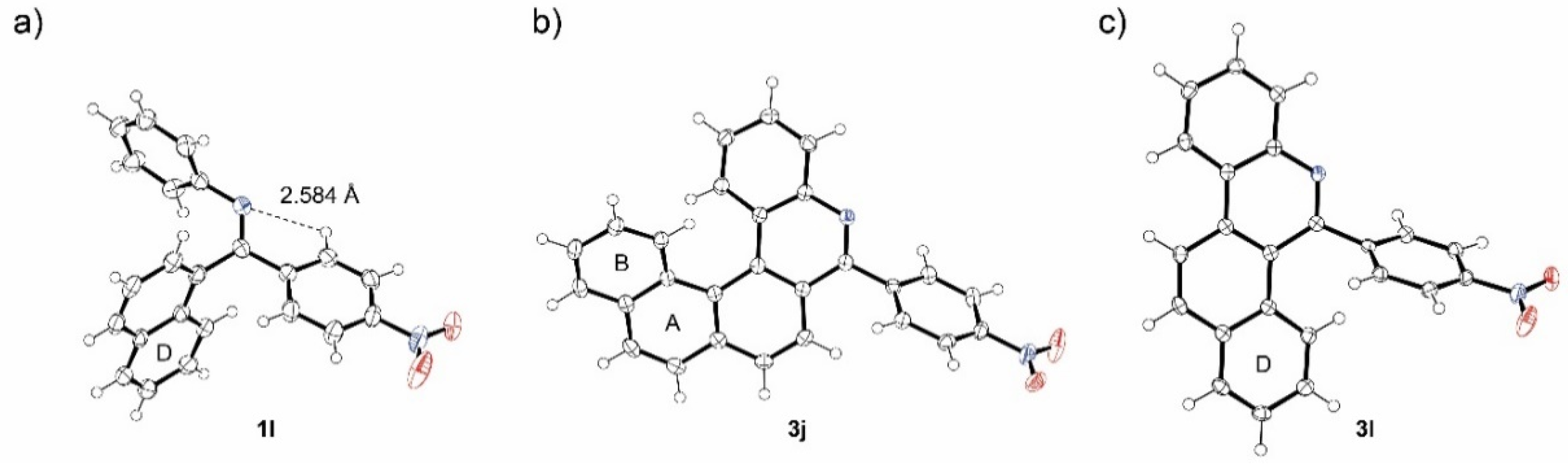
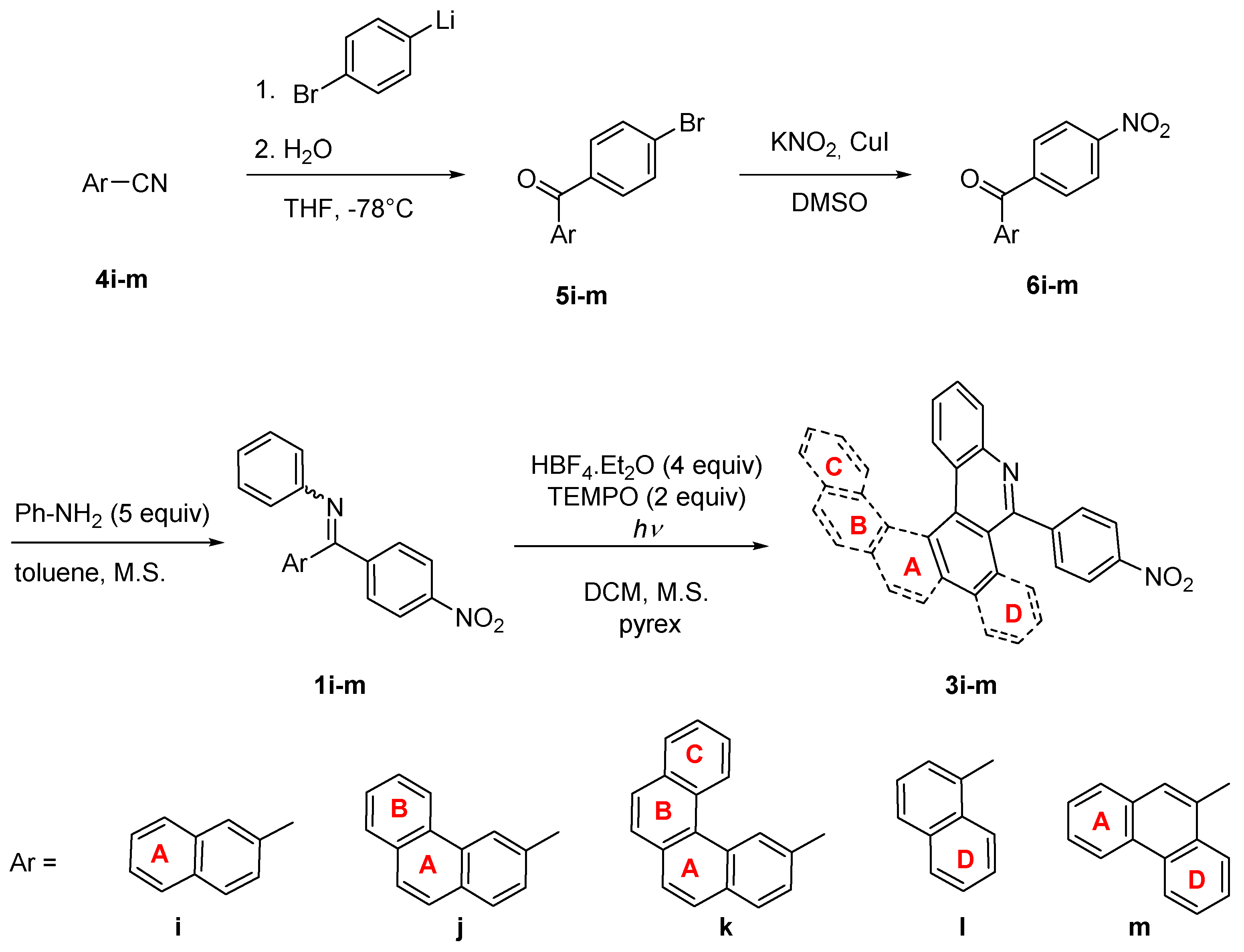
| Starting Material | Reaction Time [h] | Conversion a [%] | Isolated Yield 3i-m [%] |
|---|---|---|---|
| 1i | 10 | 100 | 36 |
| 1j | 9 | 100 | 50 |
| 1k | 13 | 0 | 0 |
| 1l | 10 | 100 | 42 |
| 1m | 10 | 100 | 40 |
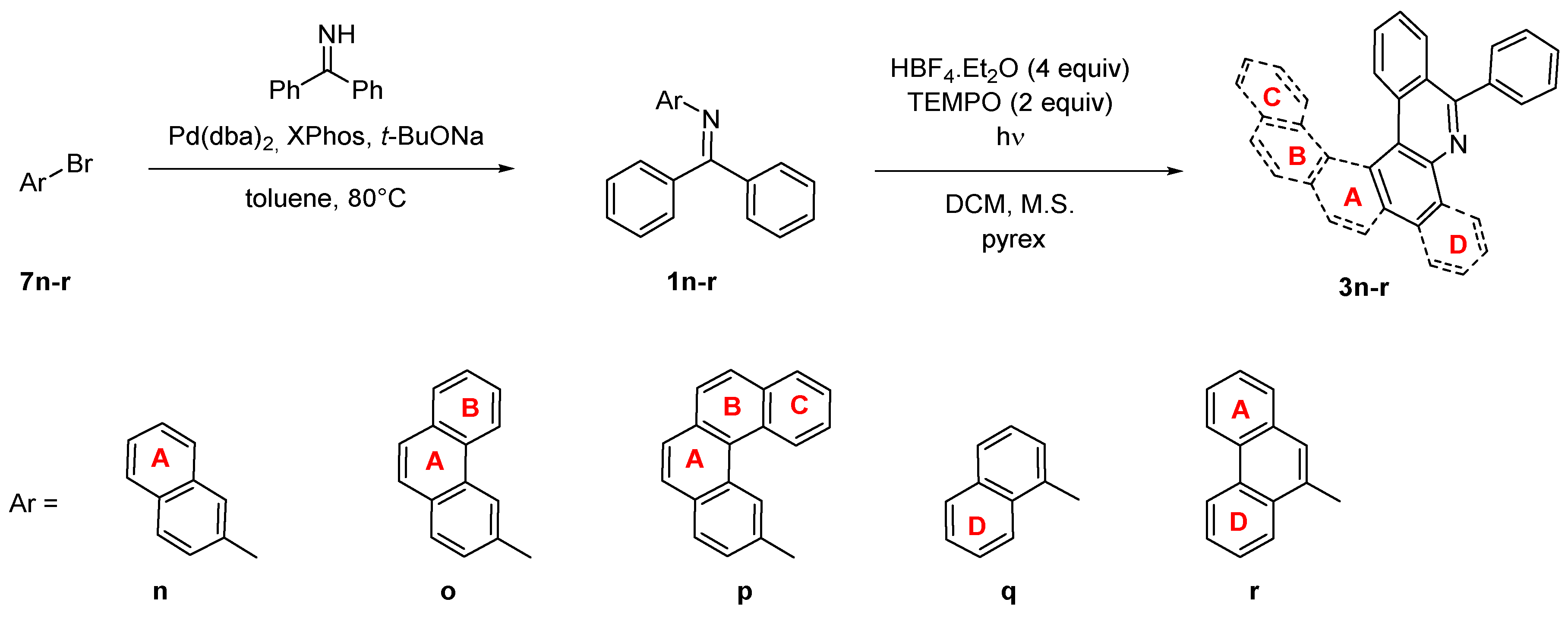
| Starting Material | Reaction Time [h] | Conversion a [%] | Isolated Yield 3n-r [%] |
|---|---|---|---|
| 1n | 8 | 100 | 64 |
| 1o | 31 | 100 | 72 |
| 1p | 22 | 0 | 0 |
| 1q | 28 | 40 | 24 |
| 1r | 15 | 100 | 72 |
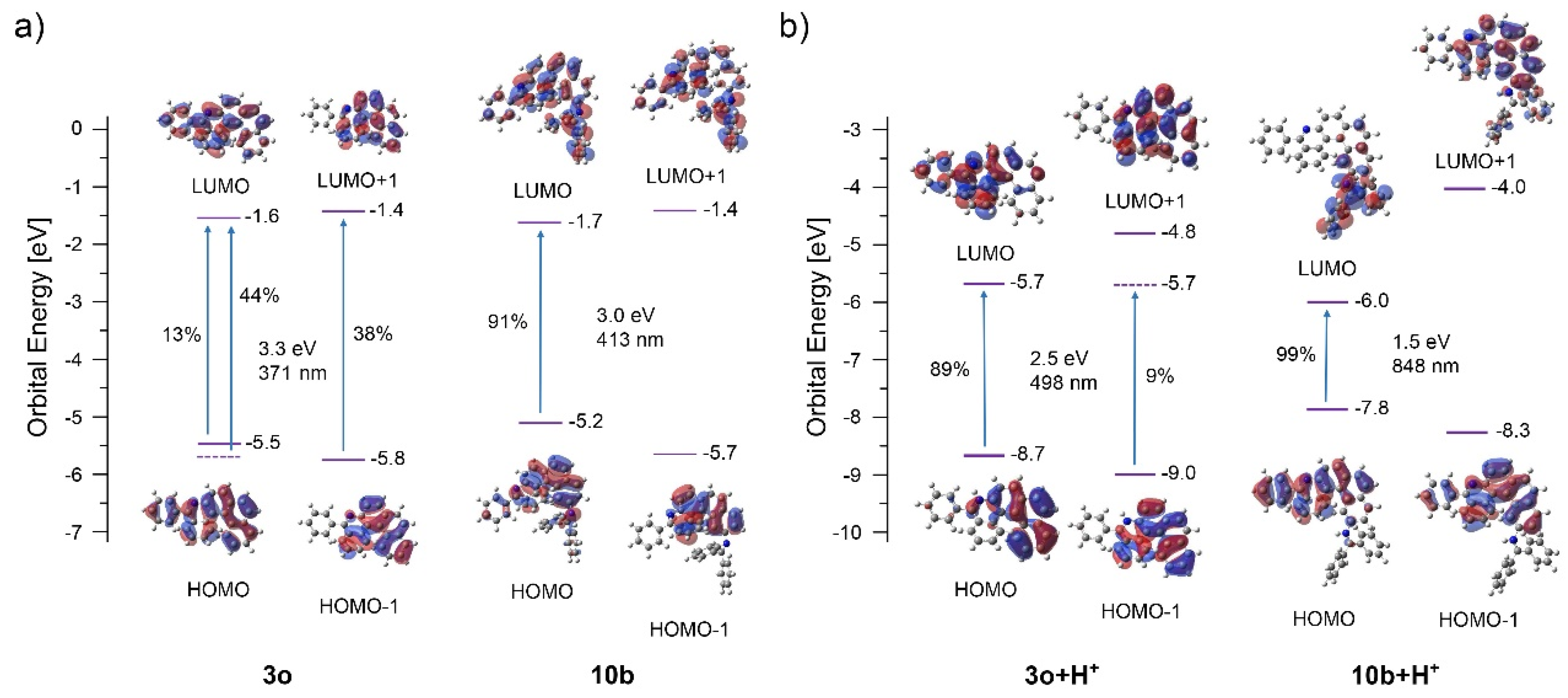
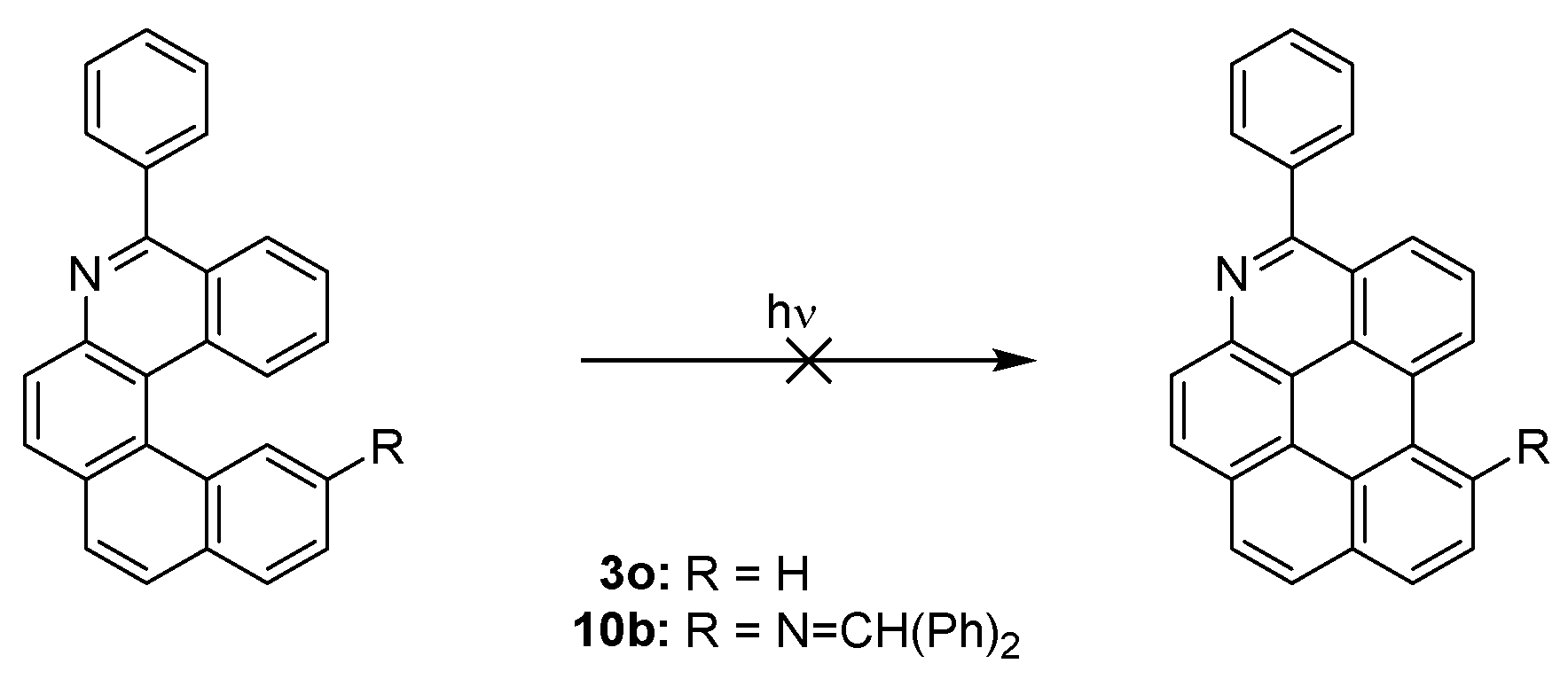
| Compound | Reaction Time [h] | Isolated Yield of 10a,b [%] | Isolated Yield of 11a,b [%] |
|---|---|---|---|
| 9a | 8 | 62 | 0 |
| 9b | 40 | 36 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kos, M.; Žádný, J.; Storch, J.; Církva, V.; Cuřínová, P.; Sýkora, J.; Císařová, I.; Kuriakose, F.; Alabugin, I.V. Oxidative Photocyclization of Aromatic Schiff Bases in Synthesis of Phenanthridines and Other Aza-PAHs. Int. J. Mol. Sci. 2020, 21, 5868. https://doi.org/10.3390/ijms21165868
Kos M, Žádný J, Storch J, Církva V, Cuřínová P, Sýkora J, Císařová I, Kuriakose F, Alabugin IV. Oxidative Photocyclization of Aromatic Schiff Bases in Synthesis of Phenanthridines and Other Aza-PAHs. International Journal of Molecular Sciences. 2020; 21(16):5868. https://doi.org/10.3390/ijms21165868
Chicago/Turabian StyleKos, Martin, Jaroslav Žádný, Jan Storch, Vladimír Církva, Petra Cuřínová, Jan Sýkora, Ivana Císařová, Febin Kuriakose, and Igor. V. Alabugin. 2020. "Oxidative Photocyclization of Aromatic Schiff Bases in Synthesis of Phenanthridines and Other Aza-PAHs" International Journal of Molecular Sciences 21, no. 16: 5868. https://doi.org/10.3390/ijms21165868