Polymorphic Forms of Valinomycin Investigated by NMR Crystallography


Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovsky Square #2, 16206 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(14), 4907; https://doi.org/10.3390/ijms21144907
Submission received: 16 June 2020
/
Revised: 8 July 2020
/
Accepted: 9 July 2020
/
Published: 11 July 2020
(This article belongs to the Special Issue Molecular Structure and Dynamics Probed by Spectroscopic Techniques and Computational Approaches: New Trends by NMR, FTIR, Neutron Scattering and Simulation)
Abstract
:A dodecadepsipeptide valinomycin (VLM) has been most recently reported to be a potential anti-coronavirus drug that could be efficiently produced on a large scale. It is thus of importance to study solid-phase forms of VLM in order to be able to ensure its polymorphic purity in drug formulations. The previously available solid-state NMR (SSNMR) data are combined with the plane-wave DFT computations in the NMR crystallography framework. Structural/spectroscopical predictions (the PBE functional/GIPAW method) are obtained to characterize four polymorphs of VLM. Interactions which confer a conformational stability to VLM molecules in these crystalline forms are described in detail. The way how various structural factors affect the values of SSNMR parameters is thoroughly analyzed, and several SSNMR markers of the respective VLM polymorphs are identified. The markers are connected to hydrogen bonding effects upon the corresponding (13C/15N/1H) isotropic chemical shifts of (CO, Namid, Hamid, Hα) VLM backbone nuclei. These results are expected to be crucial for polymorph control of VLM and in probing its interactions in dosage forms.

1. Introduction
Intense efforts are ongoing to identify treatments of the 2019 novel coronavirus (2019-nCoV) [1]. For an obvious reason, it is a very quickly evolving field that was already reviewed [2,3,4]. Most recently, a press release [5] appeared that outlined the potential of valinomycin (VLM; a cyclodepsipeptide [6] described below) in a treatment of the 2019-nCoV. This potential is assumed on the basis of the most recent report of an efficient preparation of VLM [7] and on its well-known antiviral activity [8]. We thus surmise that VLM may indeed become a part of some 2019-nCoV drug formulation(s) and investigate crystalline phases of VLM. Their description furthers the understanding of the polymorphism of VLM (the ability of a solid VLM to form various crystal modifications). The polymorphism of VLM is expected to be extensive (see the recent study of a related problem of complexation-induced structural changes in VLM [9], and references cited therein) and important for the design and manufacturing of a drug formulation. Due to a great significance of the polymorphism for pharmaceutical industry, there is a number of methods dealing with various aspects of this problem [10]. One of the approaches to the description of polymorphic forms of drug compounds is an application of the NMR crystallography [11] in its variant, most recently reviewed in reference [12], which combines the solid-state NMR (SSNMR) and computational methods together with an input from the X-ray diffraction (XRD) measurements. This NMR crystallography variant is employed in the present study. It applies the plane-wave density-functional theory (PW DFT)-based calculations of the structural and spectral parameters [13] in order to describe several crystal modifications of VLM. Two of those solid phase structures, namely, the triclinic and monoclinic forms, both featuring so called “asymmetric bracelet” conformation of the backbone (see references [14,15], respectively), were previously characterized by the SSNMR [16]. They are used for a comparison of the PW DFT and experimental results. On the basis of this comparison, two additional crystal geometries of VLM are investigated. The first one is called “symmetrical”, as it is predicted from the XRD structure of VLM—K+ complex [17], whose shape resembles a circular bracelet, after a removal of the counterions. The second predicted geometry exhibits the “propeller” structural motif (bearing resemblance to a three-blade propeller), and is obtained from the XRD structure of dimethyl sulfoxide solvate of VLM [18]. For all the aforementioned structures, their intermolecular interactions; the key internal coordinates including parameters of hydrogen bonds; and selected SSNMR data are discussed. These results should be useful also in analyses of other pharmaceutically active cyclic peptides like cyclosporins [19,20].
2. Results
2.1. General Considerations
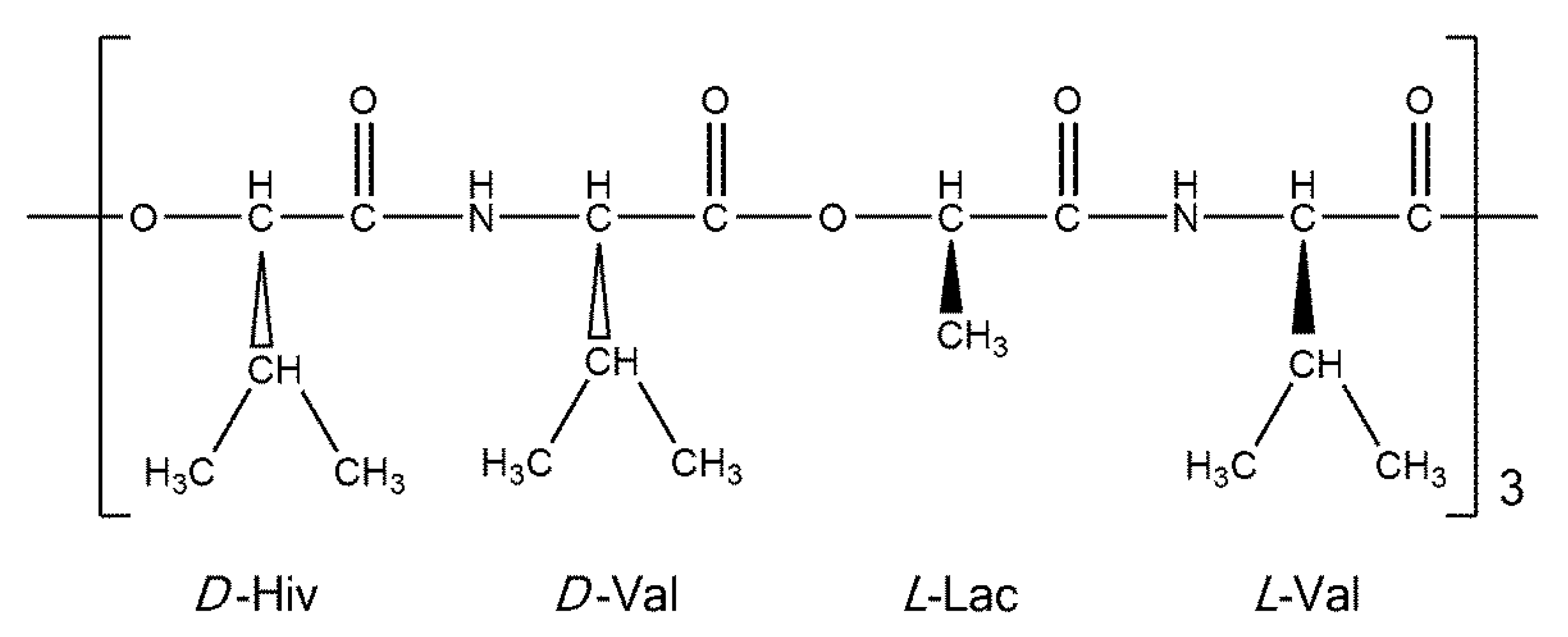
Cyclo(D-α-hydroxyisovaleryl-D-valyl-L-lactoyl-L-valyl-D-α-hydroxyisovaleryl-D-valyl-L-lactoyl-L-valyl-D-α-hydroxyisovaleryl-D-valyl-L-lactoyl-L-valyl) (valinomycin, VLM; CAS number 2001-95-8) is a cyclic dodecadepsipeptide with the ring closed at an ester linkage (see Figure 1). It contains three repetitions of (D-Hiv–D-Val–L-Lac–L-Val) segment in a macrocycle. Due to an alternation of amide and ester linkages throughout the VLM backbone, we define the backbone dihedral angles φ and ψ as follows. A quintuple of consecutive atoms {X, CO, Cα, Y, Z} is used: X denotes either amidic nitrogen (Namid) or ester oxygen of the preceding residue in the twelve-membered ring; CO and Cα are the carbonyl and alpha carbons, respectively, of the residue in question; Y denotes either Namid or ester oxygen of the residue in question; and Z denotes the carbonyl carbon of the subsequent residue (the ordering and atom numbers are specified in the Supporting Materials Tables S1–S4). Then φ is the X, CO, Cα, Y torsion angle, while ψ is the CO, Cα, Y, Z torsion angle. The [φ, ψ] values are shown below for all the investigated structures.
Importantly, none of the PW DFT optimized periodic structures (see Materials and Methods) of VLM contains any typical O…H–O or O…H–N intermolecular hydrogen bonds. For those structures, the dimerization energy, ΔE, of the closest neighboring structural units in a given VLM crystal was obtained by means of the RI-MP2/TZVP supermolecular interaction energy calculations which are described in Section 4. These complexation energies were found to be small for all four polymorphs: ΔE values amount to −22.7, −26.5, −19.4, and −36.1 kJ/mol for the dimers taken from the triclinic, monoclinic, “symmetric” and “propeller” crystal structures, respectively. They are thus comparable to the interaction energy of, for instance, an N-methylacetamide–dimethylformamide (NMA–DMF) dimer (ΔE = −26.9 kJ/mol for the PBE/aug-cc-pVQZ minimum featuring a typical hydrogen bond between the amide group of NMA and the formyl oxygen of DMF). An accuracy of the ΔE data is discussed in Section 3.
The NMR isotropic chemical shielding as obtained from the GIPAW calculations (see Materials and Methods) was converted to an estimate of the isotropic chemical shift according to references [21] (13C and 1H) and [22] (15N). These estimates are shown in Table 1, Table 2, Table 3 and Table 4 and discussed below, while the MAGRES files are obtainable from the corresponding author upon request. The tables also describe an arrangement of hydrogen bonds in the respective structures by designating, wherever applicable, a residue as the acceptor (“a”) and/or as the donor (“d”) involved in hydrogen bonding. The actual values of internal coordinates can be inferred from the coordinates which are provided, in PDB format files, in the Supporting Materials.
2.2. The Triclinic Polymorph
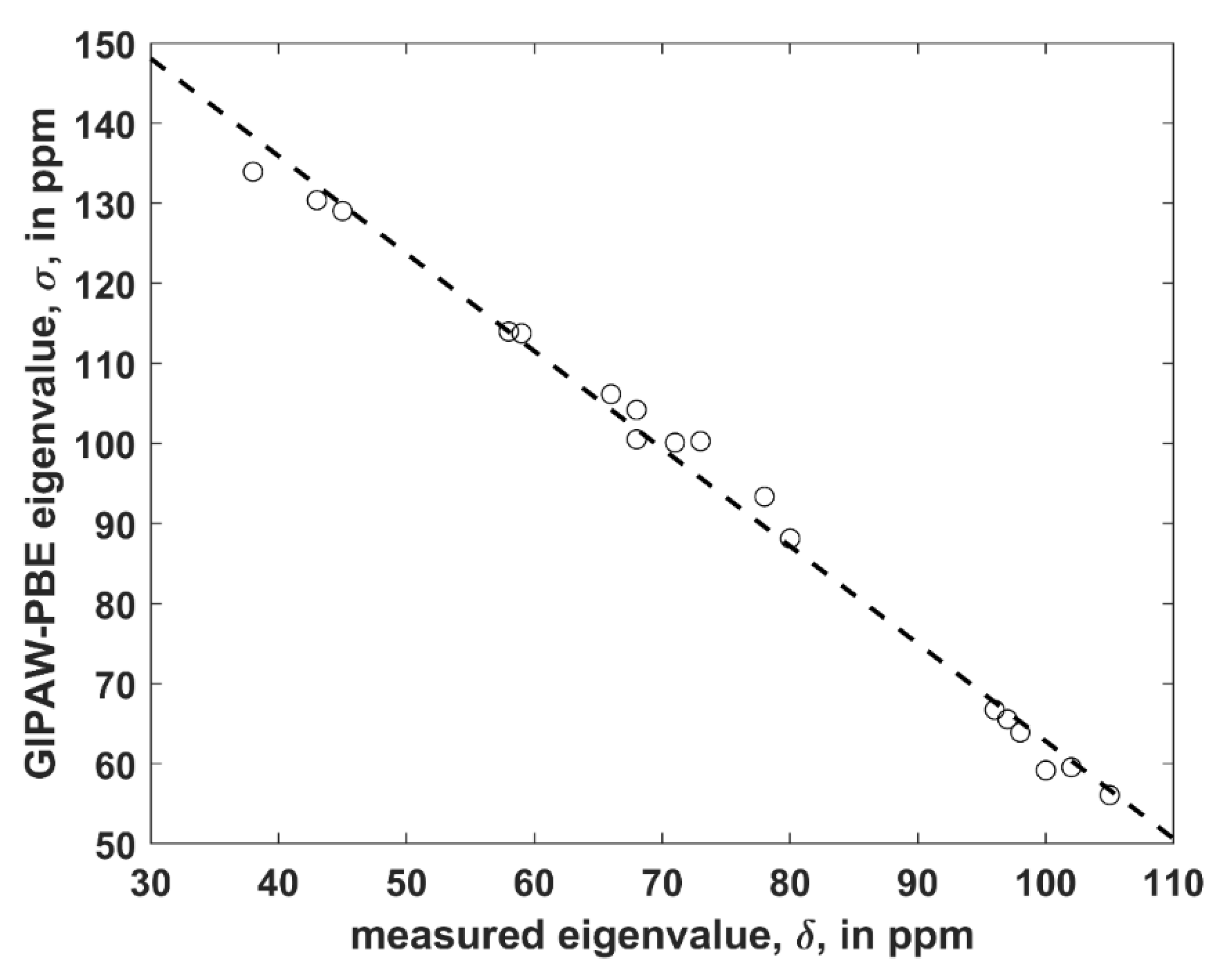
The structure was considered that is uncomplexed and crystallizes in P1 space group with two crystallographically distinct VLM molecules (further referred to as “A” and “B”; an identification of either unit in the Supporting Materials file TRICLINIC.PDB is possible through the numbering provided in Table S1). It was employed to establish a level of agreement between the PW PBE-predicted values of the principal elements of the 13C chemical shielding tensors and their measured counterparts, namely, the principal elements of the 13C chemical shift tensors. Only α carbons of D-Hyv and L-Lac sites were considered, as all six corresponding peaks were identified in the 13C SSNMR spectrum (the region of carbonyl carbons was not fully resolved) [16]. However, it should be noted that the explicit assignment of those peaks was not obtained by Kameda et al. [16] and hence it was taken from the calculations. It should also be noted that averaged values for molecules “A” and “B” were provided by the experiment [16]. Consequently, the computed results for molecules “A” and “B” were also averaged (raw data before averaging are shown in Table S5). Considering known limitations of PW DFT predictions of the principal components of the 13C chemical shielding tensors [23] and uncertainties of SSNMR measurements, the agreement between theory and experiment can be considered to be very good. Namely, a simple linear regression model (σ = a × δ + b, where σ and δ is the shorthand notation for properly ordered values of the principal elements of the 13C chemical shielding and chemical shift tensors, respectively, a is the slope, and b is the intercept; see Figure 2 has an adjusted R2 value of 0.9915, standard deviation 2.3 of ppm, and a maximum deviation of 4.6 ppm. This level of accuracy is comparable to ca. 2 ppm error of an extraction of the eigenvalues by fitting of the 13C SSNMR spectrum [16].
The triclinic polymorph features a remarkable pattern of six hydrogen bonds (see Table 1). While this pattern is well-known [14], it is investigated by the PW PBE calculations and described in detail in order to facilitate a search for the SSNMR markers identifying the polymorphs of VLM. Four of the hydrogen bonds are of the 1 → 4 type. Using “a → d” notation to specify an acceptor—donor pair, these contacts are: D-Hyv1 → L-Val4, L-Lac11 → D-Val2, D-Hyv5 → L-Val8, and L-Lac7 → D-Val10. Two hydrogen bonds are of the 1 → 5 type, namely, D-Val2 → D-Val6 and L-Val8 → L-Val12. In general, the {13C, 15N, 1H} chemical shifts of the nuclei involved in hydrogen bonding (CO, Namid, Hamid) reflect the strength of CO…Hamid–Namid contacts (see reference [24] for the most recent review of an influence of noncovalent interactions upon the SSNMR parameters). For these (CO, Namid, Hamid) nuclei, values of their corresponding {13C, 15N, 1H} chemical shifts would be expected to rise with a decreasing length of the hydrogen bond (denoted as R; a value of R is given by the distance between CO and Namid) if all other factors were equal (such an idealized dependence can be seen in Figure S1 that shows the 15Namid data of the aforementioned NMA–DMF dimer). Of course, in a real system, there are many additional contributions influencing the chemical shift value. They stem from details of an angular dependence of the hydrogen bond; a presence of the secondary structural elements; a specific conformation of the backbone; and other effects. Nevertheless, for the triclinic polymorph, it follows from an inspection of predicted chemical shielding and a concomitant analysis of the geometry that the related chemical shifts increase with shortening of the hydrogen bond. For instance, in the molecule “B” R = 320 pm for D-Val6 → D-Val2, while the relevant {13C, 15N, 1H} chemical shifts are accordingly 175.9, 114, and 8.6 ppm. An analogous fragment in the molecule “A” has a significantly lower R of 300 pm, and higher {13C, 15N, 1H} chemical shifts amounting to 176.4, 123, and 9.2 ppm, respectively. This structural difference would thus result in a clear separation of the most downfield peak at 123 ppm from the rest of the signals in the 15N SSNMR spectrum (see Table 1). As a consequence, this peak could be utilized in distinguishing between the two crystallographically independent molecules in order to establish heteronuclear correlations for the full signal assignment of the triclinic polymorph.
2.3. The Monoclinic Polymorph
The P21 structure of the monoclinic crystal form contains two symmetry-related molecules in the unit cell (visualized in the Graphical Abstract) together with one disordered n-octane molecule whose coordinates are not available [15]. Hence, only the corresponding uncomplexed VLM geometry was investigated here. It is available in the Supporting Materials file MONOCLINIC.PDB. Table 2 summarizes the predicted values of the SSNMR data and of the backbone dihedrals angles along with a specification of hydrogen bonding in this polymorph. Its packing and conformational features are analogous to both “A” and “B” molecules of the triclinic crystal structure described in the preceding paragraph. The monoclinic polymorph served to check the performance of the PW-PBE calculations for predictions of the 13C isotropic chemical shielding, σiso, of α carbons in hydroxy acid residues, as their isotropic chemical shifts, δiso, are available and were assigned to either D-Hyv or L-Lac by Kameda et al. (it is noted that one of the experimental peaks was affected by an occluded n-octane) [16]. They were fully assigned using the computed data (details are provided in Table S5). The model (see Section 2.2) σiso = −0.9452 × δiso + 165.2 ppm has an adjusted R2 value of 0.9008, standard deviation of 1.3 ppm, and a maximum deviation of 1.8 ppm. Importantly, the results reliably capture the dependence of the SSNMR data upon the solid-phase geometry of VLM, and their accuracy is at the level currently expected in NMR crystallography [25]. Thus, the PW-PBE calculations are applied with confidence to other nuclei and polymorphs examined in this work.
Based on a careful comparison of the notation used in references [14,15], a consistent numbering of the sites is adopted in Table 1 and Table 2. In the monoclinic polymorph, hydrogen bonds of the 1 → 4 type (sometimes they are used to describe “β-turn” motif in VLM structures, in analogy with the stabilized β-turns in proteins and polypeptides [26]) are D-Hyv1 → L-Val4, L-Lac3 → D-Val6, L-Lac7 → D-Val10, and D-Hyv9 → L-Val12. The 1 → 5 type hydrogen bonding is now D-Val2 → D-Val10 and L-Val4 → L-Val8. In spite of a fairly high degree of similarity between the VLM molecules of both polymorphs, values of the corresponding SSNMR parameters vary significantly (see Table 1 and Table 2) and reflect their sensitivity to seemingly small differences in the crystal packing and local effects. In particular, the hydrogen bond distance relevant for the D-Val2 → D-Val10 contact becomes R = 321 ppm and is almost the same as in the case of D-Val6 → D-Val2 in the “B” molecule of the triclinic polymorph (R = 320 pm as discussed in Section 2.2). However, due to a number of specific contributions to the chemical shifts of related (CO, Namid, Hamid) nuclei, the {13C, 15N, 1H} values are now 172.7, 116, and 7.2 ppm, respectively, to be compared to 175.9, 114, and 8.6 ppm accordingly predicted for the triclinic polymorph.
2.4. The “Symmetric” Structural Motif
Due to the known tendency of VLM to polymorphism, two additional forms occurring in complexes were modeled, which should be helpful in their future studies. The XRD structure of a VLM complex with potassium cations, that is actually an adduct of VLM, KI3, and KI5 (see reference [17]), was considered. In the complex, VLM molecules are of a high (approximately S6) symmetry, and hence this polymorph is referred to as “symmetric” here. Its geometry was obtained by removing counterions and reoptimizing the unit cell parameters together with internal coordinates. The reoptimization resulted in only a small decrease of the unit-cell volume: formally, the unit cell shrank from 15.443 nm3 [17] to 15.134 nm3, but in fact, the corresponding primitive cell of the C2221 space group was used to make computations feasible, and the actual size was one-half of those values (further details are provided in Table S11). The resulting VLM geometry, included in the Supporting Materials as SYMMETRIC.PDB file, contains six hydrogen bonds of the 1 → 4 type. On the basis of a scrutiny of the backbone dihedral angles we suppose that the VLM ring is numbered consistently throughout Table 1, Table 2, Table 3 and Table 4 in order to aid in a comparison of the predicted SSNMR data. In the present case, the hydrogen bonds are D-Hyv1 → L-Val4, L-Lac11 → D-Val2, L-Lac3 → D-Val6, D-Hyv5 → L-Val8, L-Lac7 → D-Val10, and D-Hyv9 → L-Val12. It should be realized that despite a quasisymmetric shape of a VLM molecule in this conformation, there is a non-negligible variation in parameters of those six hydrogen bonds. Specifically, the values of R lie in an interval from 285.7 to 305.1 pm with a mean of 294.8 ppm and a standard deviation of this mean amounting to 7.9 pm. This variation is in turn manifested in differences between values of the {13C, 15N, 1H} chemical shifts of the (CO, Namid, Hamid) nuclei involved in hydrogen bonding (see Table 3).
2.5. The “Propeller” Structural Motif
The last intramolecular hydrogen-bonded conformation of VLM investigated here is the one found in its crystals containing also dimethyl sulfoxide (DMSO) molecules [18]. It resembles a shallow dish with isopropyl groups at the exterior. However, due to an approximate threefold rotation symmetry and three related bends, it can be likened to a three-blade propeller as well, and is referred to simply with “propeller” (see Figure 3 that shows one of the “blades” of such a “propeller”). This structure was investigated in an analogous way as the “symmetric” polymorph, namely, by removal of coordinates of solvent molecules from the available XRD structure and the reoptimization of the crystal unit-cell together with internal coordinates of VLM, which was followed by the prediction of the SSNMR parameters. Interestingly, a reduction of the unit-cell volume was small also in the case of the “propeller” polymorph, namely, the volume decreased from 7.897 to 7.642 nm3 (the resulting unit-cell parameters are provided in Table S11, while the VLM geometry in PROPELLER.PDB file). There are only three typical hydrogen bonds in this structure: L-Lac11 → D-Val2, L-Lac3 → D-Val6, and L-Lac7 → D-Val10, with R = 298, 301, and 312 pm, respectively. The hydrogen bonding affects the 1H chemical shifts of Hamid nuclei of amino acid residues in an expected way, namely, the values are much higher in D-valines (forming hydrogen bonds) than in L-valines (with amidic protons pointing in a direction where the nearest DMSO would be in the solvate structure [18], but where there is no acceptor in the present structure). However, the 15N chemical shifts of Namid nuclei show an unanticipated pattern: the values are higher for L-valines than for hydrogen-bonded D-valines. An inspection of the 15N data in Table 1, Table 2, Table 3 and Table 4 reveals that those three D-valine nitrogens have in fact the lowest chemical shifts from among all Namid sites investigated here. This uncommon situation is addressed in the next section by describing results of a simple model that includes an influence of isopropyl sidechains upon the 15N chemical shielding. In addition, the Namid–Hamid bond lengths, the eigenvalues and orientations of the 15N chemical shielding tensors, and the 14N isotropic quadrupolar interactions of valine residues in the “propeller” polymorph are discussed below. Also of note are the 1H chemical shifts of α protons of L-Lac residues in this structure. Their values are unusually high (in the 7 ppm range, see Table 4) and are discussed in terms of the C–H…O interactions.
3. Discussion
In the previous section, four solid-phase geometries of VLM together with corresponding sets of the {13C, 15N, 1H} chemical shifts of backbone nuclei are characterized by means of the PW DFT calculations with a main goal of describing the SSNMR markers of the respective polymorphs. It should also be mentioned that the known absence of intermolecular hydrogen bonds in the crystal packing of VLM is manifested in small values of dimerization energies of the nearest molecules. Those values are found to be similar to a ΔE of the model NMA–DMF system featuring just a single intramolecular hydrogen bond (see Section 2.1 for details). Due to the size of VLM dimers (1200 electrons), only a relatively small basis set (namely, TZVP, whose use resulted in an application of 4044 basis functions anyway) was employed in RIMP2 calculations, and their accuracy could not be assessed directly. However, it is worth the effort to compare the RIMP2/TZVP ΔE to an estimate of the corresponding complete basis-set limit (CBS) value of the aforementioned NMA–DMF dimer. The aug-cc-pVXZ family of basis sets was applied to compute the underlying RIMP2 energies, and the RIMP2/CBS ΔE was approximated using the mixed Gaussian/exponential form (see Materials and Methods). The resulting −32.9 kJ/mol is 122% of −26.9 kJ/mol obtained with the TZVP basis set. It is thus safe to assume that more than one half of a total dimerization energy of VLM was recovered by the RIMP2/TZVP calculations.
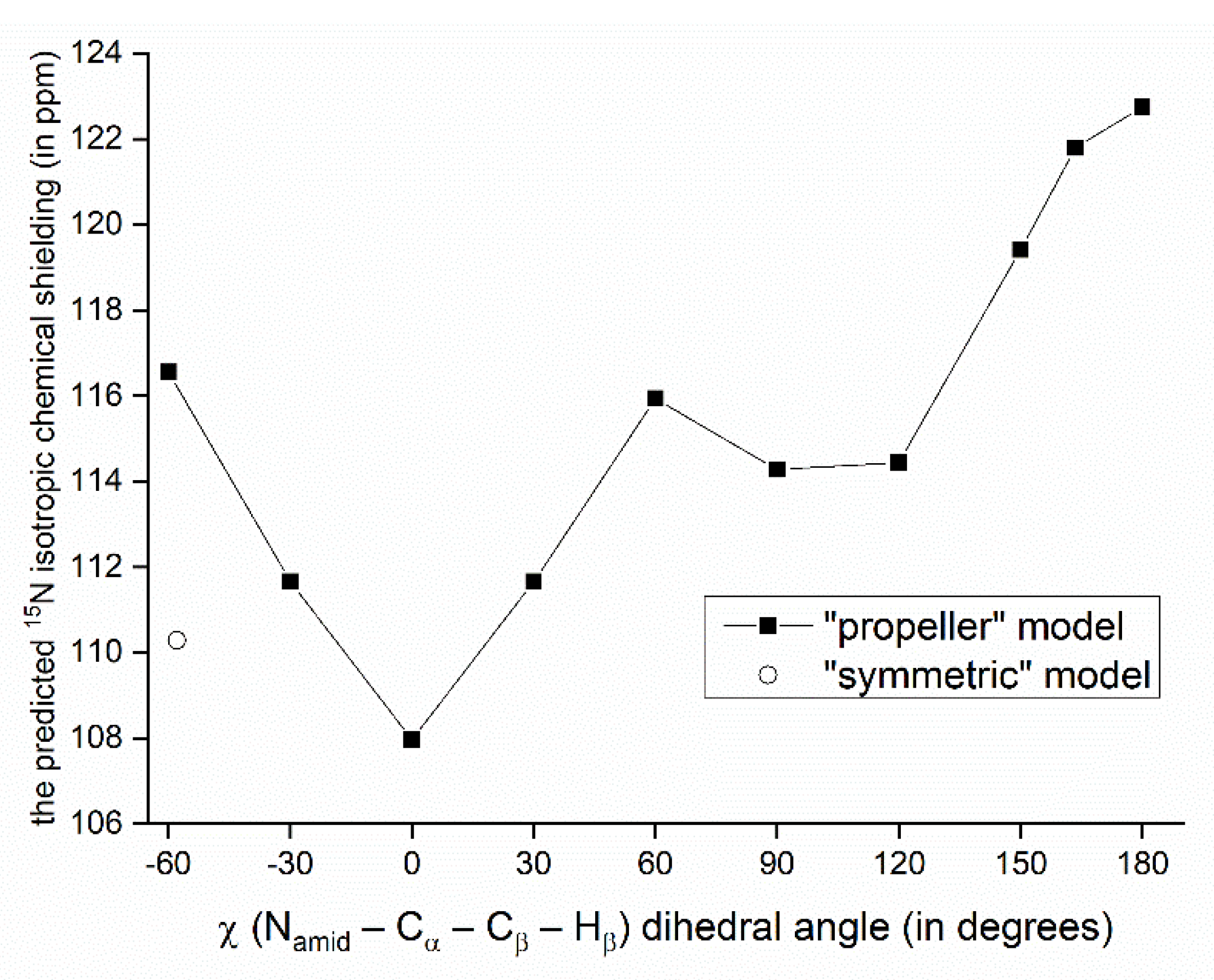
The relationship between the SSNMR data of backbone nuclei and the solid-phase geometry of VLM conformations is explored in terms of parameters of the O…Hamid–Namid hydrogen bonding. Such an analysis is not straightforward due to several other contributions to the chemical shift of some specific nucleus. In particular, the 15N chemical shifts of Namid nuclei of valine residues in the “propeller” structural motif do not follow the trend of an increase of the chemical shift value with weakening the hydrogen bond: the nitrogens of D-valines, which are hydrogen-bonded, are shielded more than those in L-valines. This shielding effect leads to unusually low values of the 15N chemical shift (predicted to be ca. 100 ppm which is to be compared to <113; 123> ppm interval of values in other polymorphs), and can be traced to an orientation of isopropyl sidechains with respect to amidic nitrogens of various structures. Specifically, in the case of D-valines in the “propeller” structure, a close contact occurs between their Namid and one of Cγ carbons of the D-valine sidechain, and protons attached to that Cγ. Since in other VLM structures the amidic nitrogens do not appear to come into such contact with neighboring sidechains, we assume that this interaction is mainly responsible for observed differences in the shielding. Our interpretation is supported by the model of a variation of the 15N chemical shielding with the sidechain dihedral angle, χ (see Figure 3 and Figure 4). This model was created from the PW PBE geometry of the “propeller” polymorph and was used to vary values of χ in an interval from −60° to +180°. For a total of 11 points from that interval, the 15N chemical shielding was obtained by the GIAO-B3LYP method (see Section 4 for details, and Table S8 for raw data) and plotted in Figure 4. It shows that Namid sites become strongly shielded at high values of χ (which is the case of D-valines in the “propeller” structure, and is of course consistent with low values of their chemical shifts). It also shows that for a χ corresponding to D-Val2 in the “symmetric” polymorph, the 15N chemical shielding is relatively low. This is consistent with a high (122 ppm) 15N chemical shift value at that site, and corroborates our structural interpretation. Nevertheless, some other parameters related to hydrogen bonding in the “propeller” structural motif were also evaluated and are summarized in Table S9. As expected, [22], the Namid–Hamid bond lengths and the 15N chemical shielding anisotropies of Namid nuclei are higher in D-valines (hydrogen-bonded residues) than in L-valines. The same trend holds for the 14Namid isotropic quadrupolar shift [27], and also for an angle between the Namid–Hamid bond vector and an eigenvector associated with the least shielded component of the 15Namid chemical shielding tensor. Values of that angle are higher by ca. 5° in D-valines as compared to L-valines (see Table S9). Moreover, since the 17O SSNMR can be usefully employed in studies of pharmaceutical compounds [28] and of hydrogen bonds [29], the related data for carbonyl oxygens are collected in Table S10.
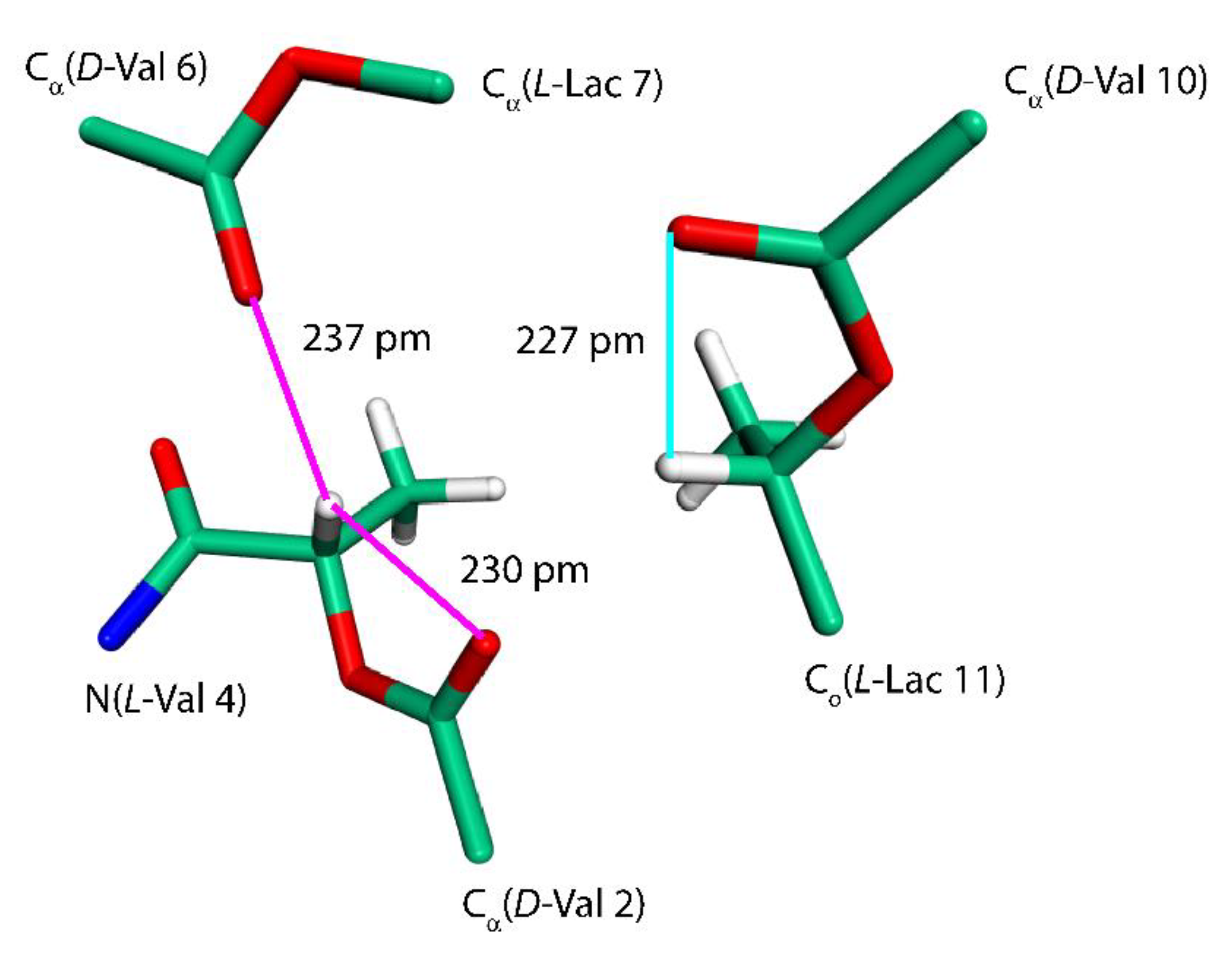
Further insight into the values of some of the SSNMR parameters can be obtained by considering an effect of C–H…O hydrogen bonds [30]. We remark that the C–H…O contacts are generally weaker than typical hydrogen bonds (for instance, an NMA–DMF dimer featuring the hydrogen bond between the carbonyl oxygen of NMA and the formyl group of DMF has, in the PBE/aug-cc-pVQZ minimum, the RIMP2/CBS ΔE value of −26.5 kJ/mol that is to be compared to −32.9 kJ/mol discussed above). However, there are numerous sites at both amino and hydroxy acid residues of VLM acting as either an acceptor or a donor involved in the respective C–H…O interactions, which thus significantly contribute to the conformational stabilization. These interactions need to be taken into account in order to be able to explain strongly downfield shifts of L-Lac Hα nuclei in the propeller polymorph (see Table 4 and Figure 5). Interestingly, the Hα of L-Lac3 with the highest chemical shift value (7.3 ppm) participates in two C–H…O hydrogen bonds (marked by the magenta lines in Figure 5). Those hydrogen bonds are slightly shorter than a single C–H…O interaction involving Hα of L-Lac11 (the cyan line in Figure 5). In any case, these chemical shifts are expected to be an important SSNMR marker of this polymorph.
The strength of the SSNMR spectroscopy lies in its ability to specify the number of crystallographically independent molecules in the unit cell and thus easily distinguish between the monoclinic and triclinic polymorphs of VLM [16]. In this study, the NMR crystallography protocol has been applied to four VLM polymorphs in order to analyze the corresponding (13C/15N/1H) isotropic chemical shifts of (CO, Namid, Hamid, Hα) backbone nuclei in terms of structural features. Should isotope-enriched VLM samples become available, other SSNMR spectral markers might include the 17O parameters [31] or 1Hamid–15Namid and 1Hα–13Cα dipolar couplings [32]. In addition, investigations of the chemical shift anisotropies could be performed [33,34,35] which would likely require measurements at very high (>20 T) magnetic fields [36,37,38].
4. Materials and Methods
The periodic PW DFT calculations were applied in the pseudopotential scheme [39,40,41] as implemented in the CASTEP 16.1 code [41]. Input files were prepared using the Materials Studio 2019 [42], and geometries were optimized with respect to the crystal-lattice energy approximated by the Perdew–Burke–Erzerhof (PBE) exchange-correlation functional [43]. In the case of computations that also optimized the crystal unit-cell parameters, the Tkatchenko–Scheffler dispersion-correction method was adopted [44]. The CASTEP calculations used the on-the-fly generated pseudopotentials and the settings consistent with “Fine” accuracy level of the Materials Studio software. Calculations were run on 60 cores of Intel® Xeon® Gold 6140 @3.70 GHz processors of the same high-performance server. We note that the optimization of the “propeller” polymorph converged exceedingly slowly: as many as 250 optimization cycles were needed to meet the convergence criteria, which took almost 18 days of CPU time of that server. For the resulting structures, the PW DFT chemical shielding tensors were predicted using the gauge-including projector augmented wave (GIPAW) method [45,46] combined with the PBE functional. The PW DFT electric-field gradient tensors were predicted, also using the PBE functional, by the method described in reference [47]. The resulting MAGRES files are obtainable.
The interaction energies were obtained from the supermolecular counterpoise-corrected calculations [48] of total energies, which were approximated by a sum of the Hartree–Fock energy and the correlation energy obtained using the resolution-of-the-identity integral approximation to the second-order Møller–Plesset energy (RIMP2) [49]. Standard triple-zeta valence plus polarization (TZVP) and augmented correlation-consistent polarized-valence basis sets (aug-cc-pVXZ, where X ∈ {D, T, Q} denotes the double-zeta (D), triple-zeta (T) and quadruple-zeta (Q) basis) were applied together with the corresponding auxiliary basis sets [50]. The RIMP2 calculations were carried out in Turbomole V7.1 [51]. The total energies were extrapolated to the complete basis set limit using an analytic solution (not shown) to the procedure described by equation 2 in reference [52].
The NMA–DMF dimers were fully optimized at the PBE/aug-cc-pVQZ level and verified to be minima of the potential energy surface by checking the values of harmonic vibrational frequencies. The chemical shielding tensors in model systems were predicted using the standard B3LYP combination of functionals applied with the standard 6-311++G(2d,2p) basis set, and with the GIAO [53,54] method to overcome the gauge problem. Additionally, for an NMA–DMF dimer the MP2-GIAO shielding tensors were predicted using the same basis set as in the B3LYP-GIAO calculations, in order to show that the chemical shielding changes are qualitatively the same (Tables S6 and S7, and Figure S1) [34]. The Gaussian 09 program package was used [55].
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/14/4907/s1. Tables S1–S4: the numbering of backbone atoms in the four polymorphs; Table S5: raw values of the SSNMR parameters of α carbons in the monoclinic and triclinic polymorphs; Tables S6 and S7: the 15Namid chemical shielding data for various intermonomer separations in an NMA–DMF dimer; Figure S1: the distance dependence of the isotropic 15Namid chemical shielding in an NMA–DMF dimer; Table S8: raw data used to create Figure 3; Tables S9 and S10: additional parameters describing the “propeller” polymorph; Table S11: the crystal unit-cell data of the four polymorphs.
Author Contributions
Conceptualization, J.C. and J.B.; Investigation, Writing, J.C.; Validation, Funding Acquisition, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
Ministry of Education, Youth and Sports of the Czech Republic project LTAUSA18011.
Acknowledgments
Access to computing and storage facilities provided under the programmes “Projects of Large Research, Development, and Innovations Infrastructures” CESNET LM2015042 and CERIT Scientific Cloud LM2015085 is greatly appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, X.; Xia, S.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. Broad-Spectrum Coronavirus Fusion Inhibitors to Combat COVID-19 and Other Emerging Coronavirus Diseases. Int. J. Mol. Sci. 2020, 21, 3843. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A. Compounds with Therapeutic Potential against Novel Respiratory 2019 Coronavirus. Antimicrob. Agents Chem. 2020, 64, e00399-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, T.-T.; Qian, J.-D.; Zhu, W.-Y.; Wang, Y.; Wang, G.-Q. A systematic review of lopinavir therapy for SARS coronavirus and MERS coronavirus-A possible for coronavirus disease-19 treatment options. J. Med. Virol. 2020, 92, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northwestern University. Super-charging drug development for COVID-19: Cell-free production method scales up yield by 5000 times. ScienceDaily. 13 April 2020. Available online: www.sciencedaily.com/releases/2020/04/200413140505.htm (accessed on 10 July 2020).
- Sivanathan, S.; Scherkenbeck, J. Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research. Molecules 2014, 19, 12368–12420. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Huang, S.; Liu, W.-Q.; Karim, A.S.; Jewett, M.C.; Li, J. Total in vitro biosynthesis of the nonribosomal macrolactone peptide valinomycin. Metab. Eng. 2020, 60, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-Q. Deciphering the Biosynthetic Codes for the Potent Anti-SARS-CoV Cyclodepsipeptide Valinomycin in Streptomyces tsusimaensis ATCC 15141. ChemBioChem 2006, 7, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.F.; Ran, X.Q.; Leitch, J.J.; Schwan, A.L.; Faragher, R.; Lipkowski, J. How Valinomycin Ionophores Enter and Transport K+ across Model Lipid Bilayer Membranes. Langmuir 2019, 35, 16935–16943. [Google Scholar] [CrossRef]
- Hilfkier, R.; Von Raumer, M. Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development; Wiley-VCH: Hoboken, NJ, USA, 2019. [Google Scholar]
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. NMR Crystallography; Wiley: Chichester, UK, 2009. [Google Scholar]
- Hodgkinson, P. NMR Crystallography of Molecular Organics. Prog. Nucl. Mag. Res. Sp. 2020, 118, 10–53. [Google Scholar] [CrossRef]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azais, T.; Asbrook, S.E.; Griffin, J.M.; Yates, J.R.; Pickard, J.C. First-Principles Calculation of NMR Parameters Using the Gauge Including Projector Augmented Wave Method: A Chemist’s Point of View. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef]
- Karle, I.L. Conformation of Valinomycin in a Triclinic Crystal Form. J. Am. Chem. Soc. 1975, 97, 4379–4386. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Duax, D.L.; Langs, D.A.; DeTitta, G.T.; Edmonds, J.W.; Rohrer, D.C.; Weeks, C.M. The Crystal and Molecular Structure of the Triclinic and Monoclinic Forms of Valinomycin, C54H90N6O18. J. Am. Chem. Soc. 1975, 97, 7242–7247. [Google Scholar] [CrossRef] [PubMed]
- Kameda, T.; McGeorge, G.; Orendt, A.; Grant, D.M. 13C NMR chemical shifts of the triclinic and monoclinic crystal forms of valinomycin. J. Biomol. NMR 2004, 29, 281–288. [Google Scholar] [CrossRef]
- Neupert-Laves, K.; Dobler, M. The Crystal Structure of a K+ Complex of Valinomycin. Helv. Chim. Acta 1975, 58, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Karle, I.L.; Flippen-Anderson, J.L. New conformation exhibiting near three-fold symmetry for uncomplexed valinomycin in crystals from dimethyl sulfoxide. J. Am. Chem. Soc. 1988, 110, 3253–3257. [Google Scholar] [CrossRef]
- Wu, K.; Gore, A.; Graham, R. A New Hydrate of Cyclosporin: Structural and Physicochemical Characterization. J. Pharm. Sci. 2018, 107, 3070–3079. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, D.; Grosjean, B.; Bodenhausen, G. Dipolar couplings in solid polypeptides probed by 14N NMR spectroscopy. Chem. Sci. 2018, 1, 1–9. [Google Scholar] [CrossRef]
- Paruzzo, F.M.; Hofstetter, A.; Musil, F.; De, S.; Ceriotti, M.; Emsley, L. Chemical shifts in molecular solids by machine learning. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Czernek, J.; Brus, J. Monitoring the Site-Specific Solid-State NMR Data in Oligopeptides. Int. J. Mol. Sci. 2020, 21, 2700. [Google Scholar] [CrossRef]
- Czernek, J.; Pawlak, T.; Potrzebowski, M.J. Benchmarks for the 13C NMR chemical shielding tensors in peptides in the solid state. Chem. Phys. Lett. 2012, 527, 31–35. [Google Scholar] [CrossRef]
- Xu, Y.; Szell, P.M.J.; Kumar, V.; Bryce, D.L. Solid-state NMR spectroscopy for the analysis of element-based non-covalent interactions. Coord. Chem. Rev. 2020, 411, 213237. [Google Scholar] [CrossRef]
- Czernek, J.; Urbanova, M.; Brus, J. NMR Crystallography of the Polymorphs of Metergoline. Crystals 2018, 8, 378. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.-M.; Kassees, K.; Lee, T.-K.; Manadhar, B.; Yousif, A.M. Strategy and Tactics for Designing Analogs: Biochemical Characterization of the Large Molecules. In Comprehensive Medicinal Chemistry, 3rd ed.; Chackalamannil, S., Rotella, E., Ward, S.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 66–115. [Google Scholar]
- Tatton, A.S.; Pham, T.N.; Vogt, F.G.; Iuga, D.; Edwards, A.J.; Brown, S.P. Probing Hydrogen Bonding in Cocrystals and Amorphous Dispersions Using 14N–1H HMQC Solid-State NMR. Mol. Pharm. 2013, 10, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.Q.; Shan, M.; Terskikh, V.; Hung, I.; Gan, Z.H.; Wu, G. Solid-State 17O NMR of Pharmaceutical Compounds: Salicylic Acid and Aspirin. J. Phys. Chem. B 2013, 117, 9643–9654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.S.; Hung, I.; Brinkmann, A.; Gan, Z.H.; Kong, X.Q.; Wu, G. Solid-State 17O NMR Reveals Hydrogen-Bonding Energetics: Not All Low-Barrier Hydrogen Bonds Are Strong. Angew. Chem. 2017, 129, 6262–6266. [Google Scholar] [CrossRef]
- Vibhute, A.M.; Priyakumar, U.D.; Ravi, A.; Sureshan, K.M. Model molecules to classify C–H…O hydrogen-bonds. Chem. Commun. 2018, 54, 4629–4632. [Google Scholar] [CrossRef] [PubMed]
- Wu, G. 17O NMR studies of organic and biological molecules in aqueous solution and in the solid state. Prog. Nucl. Mag. Res. Sp. 2019, 114, 135–191. [Google Scholar] [CrossRef]
- Awosanya, E.O.; Lapin, J.; Nevzorov, A.A. NMR “crystallography” for uniformly (13C, 15N)-labeled oriented membrane proteins. Angew. Chem. 2020, 59, 3554–3557. [Google Scholar] [CrossRef]
- Rossi, F.; Duong, N.T.; Pandey, M.K.; Chierotti, M.R.; Gobetto, R.; Nishiyama, Y. Determination of the 15N chemical shift anisotropy in natural abundance samples by proton-detected 3D solid-state NMR under ultrafast MAS of 70 kHz. Magn. Reson. Chem. 2019, 57, 294–303. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Exploring Accuracy Limits of Predictions of the 1H NMR Chemical Shielding Anisotropy in the Solid State. Molecules 2019, 24, 1731. [Google Scholar] [CrossRef] [Green Version]
- Kraus, J.; Gupta, R.; Lu, M.; Gronenborn, A.M.; Akke, M.; Polenova, T. Accurate Backbone 13C and 15N Chemical Shift Tensors in Galectin-3 Determined by MAS NMR and QM/MM: Details of Structure and Environment Matter. ChemPhysChem 2020, 21, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.M.; Wang, M.; Polenova, T. NMR of Macromolecular Assemblies and Machines at 1 GHz and Beyond: New Transformative Opportunities for Molecular Structural Biology. Methods Mol. Biol. 2018, 1688, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.; Wang, X.L.; Hung, I.; Gan, Z.H.; Gervais, C.; Sassoye, C.; Rimsza, J.; Du, J.C.; Smith, M.E.; Hanna, J.V.; et al. Pushing the limits of sensitivity and resolution for natural abundance 43Ca NMR using ultra-high magnetic field (35.2 T). Chem. Commun. 2018, 54, 9591–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulino, J.; Yi, M.; Hung, I.; Gan, Z.H.; Wang, X.L.; Chekmenev, E.Y.; Zhou, H.X.; Cross, T.A. Functional stability of water wire-carbonyl interactions in an ion channel. Proc. Natl. Acad. Sci. USA 2020, 117, 11908–11915. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First principles simulation: Ideas, illustrations, and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA Materials Studio. Dassault Systèmes, Vélizy-Villacoublay: Paris, France. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 10 July 2020).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [Green Version]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculations of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Profeta, M.; Mauri, F.; Pickard, C.J. Accurate First Principles Predictions of 17O NMR Parameters in SiO2: Assignment of the Zeolite Ferrierite Spectrum. J. Am. Chem. Soc. 2003, 125, 541–548. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–556. [Google Scholar] [CrossRef]
- Vahtras, O.; Almlof, J.; Feyereisen, M.W. Integral Approximations for LCAO-SCF Calculations. Chem. Phys. Lett. 1993, 213, 514–518. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kŏlmel, C. Electronic-Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunnig, T.J., Jr. Benchmark calculations with correlated wave functions. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculation. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Frish, M.J.; Trucks, J.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
Figure 1.
A schematic representation of valinomycin.

Figure 2.
The linear regression model of the 13C SSNMR data described in the text (the dashed line is σ = −1.218 × δ + 184.6 ppm).
Figure 2.
The linear regression model of the 13C SSNMR data described in the text (the dashed line is σ = −1.218 × δ + 184.6 ppm).

Figure 3.
The model employed to calculate changes in the 15N chemical shielding of the amidic nitrogen of D-Val2 residue (this geometry features a χ, depicted in orange, of 163°).
Figure 3.
The model employed to calculate changes in the 15N chemical shielding of the amidic nitrogen of D-Val2 residue (this geometry features a χ, depicted in orange, of 163°).

Figure 4.
Variation of the GIAO-B3LYP/6-311++G(2d,2p) 15N chemical shielding with the sidechain dihedral angle. The point marked by an open circle has of a value of χ found in the PW PBE geometry of the “symmetric” polymorph.
Figure 4.
Variation of the GIAO-B3LYP/6-311++G(2d,2p) 15N chemical shielding with the sidechain dihedral angle. The point marked by an open circle has of a value of χ found in the PW PBE geometry of the “symmetric” polymorph.

Figure 5.
The fragment of a VLM molecule of the “propeller” polymorph. See the text for details.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The PW PBE structural and spectral parameters of the triclinic crystalline phase of VLM (the values for the second crystallographically independent molecule are shown in parentheses).
Table 1.
The PW PBE structural and spectral parameters of the triclinic crystalline phase of VLM (the values for the second crystallographically independent molecule are shown in parentheses).
| Residue | H-Bonding Scheme | Corresponding {13C/15N/1H} NMR Chemical Shift (in ppm) | Dihedral Angle (in Degrees) | |||||
|---|---|---|---|---|---|---|---|---|
| CO | Cα | Namid | Hamid | Hα | φ | ψ | ||
| D-Hyv1 | a ← d4 (a ← d4) | 170.2 (171.0) | 76.6 (75.1) | – | – | 5.8 (6.2) | −2 (−7) | +94 (+102) |
| D-Val2 | d → a11, a ← d6 (d → a11, a ← d6) | 176.4 (175.9) | 60.7 (58.5) | 123 (114) | 9.2 (8.6) | 3.7 (4.8) | −124 (−135) | +65 (+64) |
| L-Lac3 | none (none) | 166.8 (166.3) | 74.9 (75.6) | – | – | 5.5 (5.2) | −11 (−11) | −68 (−68) |
| L-Val4 | d → a1 (d → a1) | 171.7 (172.3) | 54.2 (53.8) | 113 (112) | 9.1 (8.3) | 5.7 (5.9) | +68 (+76) | −109 (−107) |
| D-Hyv5 | a ← d8 (a ← d8) | 168.4 (169.6) | 78.0 (78.3) | – | – | 6.1 (6.3) | −6 (−11) | +153 (+142) |
| D-Val6 | d → a2 (d → a2) | 173.7 (173.7) | 60.2 (61.1) | 117 (116) | 7.5 (8.0) | 3.7 (3.7) | −130 (−129) | +57 (+62) |
| L-Lac7 | a ← d10 (a ← d10) | 171.8 (171.8) | 70.3 (70.0) | – | – | 6.1 (6.0) | +9 (+9) | −96 (−96) |
| L-Val8 | d → a5, a ← d12 (d → a5, a ← d12) | 177.9 (177.7) | 61.5 (61.7) | 120 (120) | 9.3 (9.5) | 4.0 (3.9) | +128 (+129) | −68 (−65) |
| D-Hyv9 | none (none) | 166.1 (166.3) | 83.4 (83.2) | – | – | 5.7 (5.6) | +3 (+4) | +82 (+81) |
| D-Val10 | d → a7 (d → a7) | 173.1 (173.3) | 55.2 (54.6) | 115 (116) | 8.5 (8.6) | 5.6 (5.9) | −66 (−66) | +106 (+104) |
| L-Lac11 | a ← d2 (a ← d2) | 171.3 (170.5) | 72.9 (72.5) | – | – | 6.7 (6.2) | +26 (+21) | −161 (−159) |
| L-Val12 | d → a8 (d → a8) | 173.7 (173.5) | 60.8 (61.1) | 116 (115) | 8.5 (8.2) | 4.0 (3.7) | +128 (+125) | −68 (−68) |
Table 2.
The PW PBE structural and spectral parameters of the monoclinic crystalline phase of VLM.
| Residue | H-Bonding Scheme | Corresponding {13C/15N/1H} NMR Chemical Shift (in ppm) | Dihedral Angle (in Degrees) | |||||
|---|---|---|---|---|---|---|---|---|
| CO | Cα | Namid | Hamid | Hα | φ | ψ | ||
| D-Hyv1 | a ← d4 | 168.9 | 76.2 | – | – | 6.3 | −7 | +142 |
| D-Val2 | d → a10 | 172.7 | 61.4 | 116 | 7.2 | 3.7 | −130 | +59 |
| L-Lac3 | a ← d6 | 171.5 | 71.1 | – | – | 6.1 | +9 | −96 |
| L-Val4 | d → a1, a ← d8 | 177.7 | 61.1 | 119 | 9.6 | 4.0 | +127 | −67 |
| D-Hyv5 | none | 165.8 | 82.7 | – | – | 5.7 | +2 | +81 |
| D-Val6 | d → a3 | 172.7 | 54.8 | 115 | 8.5 | 5.6 | −68 | +106 |
| L-Lac7 | a ← d10 | 170.0 | 70.3 | – | – | 6.3 | +22 | −155 |
| L-Val8 | d → a4 | 173.2 | 61.5 | 115 | 7.9 | 3.6 | +128 | −67 |
| D-Hyv9 | a ← d12 | 171.2 | 74.2 | – | – | 6.0 | −3 | +95 |
| D-Val10 | d → a7, a ← d2 | 176.5 | 59.3 | 122 | 9.0 | 4.0 | −125 | +66 |
| L-Lac11 | none | 166.9 | 73.6 | – | – | 5.5 | −11 | −77 |
| L-Val12 | d → a9 | 171.7 | 54.1 | 113 | 8.9 | 5.8 | +74 | −105 |
Table 3.
The PW PBE structural and spectral parameters of the “symmetric” crystalline phase of VLM.
| Residue | H-Bonding Scheme | Corresponding {13C/15N/1H} NMR Chemical Shift (in ppm) | Dihedral Angle (in Degrees) | |||||
|---|---|---|---|---|---|---|---|---|
| CO | Cα | Namid | Hamid | Hα | φ | ψ | ||
| D-Hyv1 | a ← d4 | 170.3 | 75.9 | – | – | 5.8 | +1 | +101 |
| D-Val2 | d → a11 | 173.2 | 60.8 | 122 | 9.0 | 4.2 | −123 | +66 |
| L-Lac3 | a ← d6 | 172.5 | 72.3 | – | – | 5.4 | −17 | −79 |
| L-Val4 | d → a1 | 171.3 | 58.8 | 115 | 9.5 | 4.1 | +107 | −78 |
| D-Hyv5 | a ← d8 | 169.9 | 77.4 | – | – | 6.4 | −8 | +114 |
| D-Val6 | d → a3 | 169.7 | 59.0 | 121 | 9.0 | 4.3 | −101 | +83 |
| L-Lac7 | a ← d10 | 170.4 | 69.4 | – | – | 6.2 | +8 | −125 |
| L-Val8 | d → a5 | 170.8 | 59.2 | 119 | 9.8 | 4.3 | +106 | −80 |
| D-Hyv9 | a ← d12 | 170.3 | 75.5 | – | – | 6.2 | +3 | +115 |
| D-Val10 | d → a7 | 174.4 | 60.1 | 122 | 9.4 | 4.0 | −128 | +64 |
| L-Lac11 | a ← d2 | 171.3 | 71.8 | – | – | 5.2 | −18 | −76 |
| L-Val12 | d → a9 | 171.2 | 59.7 | 114 | 8.9 | 3.9 | +118 | −70 |
Table 4.
The PW PBE structural and spectral parameters of the “propeller” crystalline phase of VLM.
| Residue | H-Bonding Scheme | Corresponding {13C/15N/1H} NMR Chemical Shift (in ppm) | Dihedral Angle (in Degrees) | |||||
|---|---|---|---|---|---|---|---|---|
| CO | Cα | Namid | Hamid | Hα | φ | ψ | ||
| D-Hyv1 | none | 167.1 | 79.0 | – | – | 6.4 | 0 | +104 |
| D-Val2 | d → a11 | 173.0 | 52.2 | 101 | 9.0 | 6.1 | −177 | +125 |
| L-Lac3 | a ← d6 | 171.9 | 70.9 | – | – | 7.3 | +19 | −146 |
| L-Val4 | none | 173.5 | 59.9 | 113 | 7.2 | 4.6 | +116 | 72 |
| D-Hyv5 | none | 165.5 | 80.0 | – | – | 6.3 | −4 | +10 |
| D-Val6 | d → a3 | 174.5 | 53.7 | 100 | 8.5 | 6.5 | −170 | +121 |
| L-Lac7 | a ← d10 | 171.3 | 70.1 | – | – | 7.0 | +35 | −142 |
| L-Val8 | none | 174.2 | 68.6 | 113 | 6.9 | 4.7 | +122 | −70 |
| D-Hyv9 | none | 166.4 | 80.2 | – | – | 6.1 | +3 | +88 |
| D-Val10 | d → a7 | 172.5 | 53.4 | 99 | 8.3 | 6.0 | −177 | +121 |
| L-Lac11 | a ← d2 | 168.8 | 71.9 | – | – | 7.0 | +25 | −137 |
| L-Val12 | none | 172.3 | 58.5 | 112 | 6.7 | 4.4 | +110 | −72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Czernek, J.; Brus, J. Polymorphic Forms of Valinomycin Investigated by NMR Crystallography. Int. J. Mol. Sci. 2020, 21, 4907. https://doi.org/10.3390/ijms21144907
AMA Style
Czernek J, Brus J. Polymorphic Forms of Valinomycin Investigated by NMR Crystallography. International Journal of Molecular Sciences. 2020; 21(14):4907. https://doi.org/10.3390/ijms21144907
Chicago/Turabian StyleCzernek, Jiří, and Jiří Brus. 2020. "Polymorphic Forms of Valinomycin Investigated by NMR Crystallography" International Journal of Molecular Sciences 21, no. 14: 4907. https://doi.org/10.3390/ijms21144907
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.