3.2.1. Preparation of Alkynylcalixarenes 5a–f
General Procedure for the Synthesis of Alkynylcalixarenes 5a–f (Method A)
Starting calix[4]arene 3 (1 equiv), CuI (0.1 equiv) and Pd(PPh3)2Cl2 (0.05 equiv) were dissolved in 3–4 mL of dry piperidine under an Ar atmosphere. Then, appropriate alkyne 4a–f (1.1 equiv) was added, and the reaction mixture was stirred and heated at 100 °C for 3 days. The mixture was cooled to RT, and the solvent was evaporated under reduced pressure. The crude product was extracted (3 × 30 mL) with dichloromethane (DCM), the organic phase was washed with water, dried over MgSO4, filtered and evaporated to dryness. The residue was purified by column chromatography on silica gel (eluent: DCM: cyclohexane) to afford the title compounds.
General Procedure for the Synthesis of Alkynylcalixarenes 5a–f (Method B)
A mixture of starting calix[4]arene 3 (1 equiv), alkyne 4a–f (4 equiv), piperidine (4 equiv), CuI (0.2 equiv), Pd(PPh3)4 (0.1 equiv) and dry THF was stirred under argon atmosphere at 70 °C for 4 h in the microwave reactor. The mixture was then evaporated to dryness, the residue was extracted with DCM (3 × 30 mL), washed with water and dried over MgSO4. The purification of the crude product was done in the same way as described in Method A.
Alkynylcalixarene 5a
Method A: Calixarene 3 (0.209 mmol, 150 mg), 3,5-dimethoxyphenylacetylene (0.230 mmol, 37 mg), CuI (0.0209 mmol, 4 mg) and Pd(PPh3)2Cl2 (0.0104 mmol, 7 mg) were used under standard conditions; the product was purified by column chromatography on silica gel (DCM/cyclohexane 1:1) to afford 5a in 89% yield (140 mg) as a white solid.
Method B: Calixarene 3 (0.139 mmol, 104 mg), 3,5-dimethoxyphenylacetylene (0.556 mmol, 90 mg), CuI (0.028 mmol, 5 mg), piperidine (0.417 mmol, 35 µL) and Pd(PPh3)4 (0.0139 mmol, 16 mg) were used under standard conditions. The product 5a (77 mg, 74%) was obtained by column chromatography on silica gel (DCM/cyclohexane 60:40) as a white solid.
Data for compound 5a: M.p. = 105–107 °C. 1H NMR (CDCl3, 500 MHz, 298 K) δ 7.19 (d, J = 7.7 Hz, 1H, Ar-H), 7.08 (d, J = 7.7 Hz, 2H, Ar-H), 7.06 (d, J = 7.7 Hz, 1H, Ar-H), 6.89 (t, J = 7.5 Hz, 1H, Ar-H), 6.67 (d, J = 2.3 Hz, 2H, Ar-H), 6.43 (t, J = 2.3 Hz, 1H, Ar-H), 6.33 (dd, J = 7.5 Hz, J = 1.1 Hz, 1H, Ar-H), 6.26 (t, J = 7.5 Hz, 1H, Ar-H), 6.24 (t, J = 7.5 Hz, 1H, Ar-H), 6.15 (d, J = 7.5 Hz, 2H, Ar-H), 6.13 (d, J = 7.5 Hz, 1H, Ar-H), 4.47 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 4.46 (d, J = 2.8 Hz, 1H, Ar-CH2-Ar), 4.44 (d, J = 2.8 Hz, 1H, Ar-CH2-Ar), 4.36 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 4.01 (m, 4H, -O-CH2-), 3.95 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 3.79 (s, 6H, -O-CH3), 3.73 (m, 2H, -O-CH2-), 3.69 (m, 2H, -O-CH2-), 3.16 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 3.15 (d, J = 13.2 Hz, 2H, Ar-CH2-Ar), 1.98 (m, overlap, 4H, -CH2-CH3), 1.88 (m, 4H, -CH2-CH3), 1.10 (t, J = 7.3 Hz, 6H, -O-(CH2)2-CH3), 0.91 (t, J = 7.4 Hz, 3H, -CH3), 0.90 (t, J = 7.4 Hz, 3H, -CH3). 13C NMR (CDCl3, 126 MHz, 298 K) δ 160.47, 157.98, 157.88, 155.17, 155.16, 139.31, 137.99, 137.01, 136.89, 133.47, 133.22, 132.92, 132.67, 128.84, 128.78, 128.51, 127.57, 127.41, 127.33, 127.24, 126.21, 125.01, 122.10 (2C), 121.82, 121.78, 109.33, 101.42, 91.1, 88.83, 76.94, 76.90, 76.58, 76.44, 55.43 (2C), 31.04, 30.97, 30.96, 28.11, 23.50 (2C), 22.99, 22.97, 10.79, 10.78, 9.86, 9.84 ppm. HRMS (ESI+) calcd for C50H56O6 775.3969 [M + Na]+, found m/z 775.3971 [M + Na]+. IR (KBr) ν 2959, 2932, 2873, 1587, 1453, 1418, 1205, 1193, 1154, 1060, 965, 756 cm−1.
Alkynylcalixarene 5b
Method A: Calixarene 3 (0.315 mmol, 226 mg), phenylacetylene (0.3465 mmol, 40 µL), CuI (0.0315 mmol, 6 mg) and Pd(PPh3)2Cl2 (0.0157 mmol, 11 mg) were used under standard conditions; the product was purified by column chromatography on silica gel (DCM/cyclohexane 1:1) to afford 5b in 96% yield (209 mg) as a yellow oil.
Method B: Calixarene 3 (0.139 mmol, 103 mg), phenylacetylene (0.556 mmol, 60 µL), CuI (0.028 mmol, 5 mg), piperidine (0.417 mmol, 40 µL) and Pd(PPh3)4 (0.0139 mmol, 16 mg) were used under standard conditions. The product 5b (59 mg, 62%) was obtained by column chromatography on silica gel (DCM/cyclohexane 70:30) as a yellow oil.
Data for compound 5b: 1H NMR (CDCl3, 500 MHz, 298 K) δ 7.51 (dd, J = 7.7 Hz, J = 1.6 Hz, 2H, Ar-H), 7.27–7.35 (m, 3H, Ar-H) 7.19 (d, J = 7.7 Hz, 1H, Ar-H), 7.08 (d, J = 7.4 Hz, 2H, Ar-H), 7.06 (d, J = 7.7 Hz, 1H, Ar-H), 6.90 (t, J = 7.7 Hz, 1H, Ar-H), 6.34 (d, J = 7.7 Hz, 1H, Ar-H), 6.26 (d, J = 7.7 Hz, 1H, Ar-H), 6.22 (d, J = 7.7 Hz, 1H, Ar-H), 6.14 (d, J = 8.2 Hz, 2H, Ar-H), 6.12 (d, J = 7.7 Hz, 1H, Ar-H), 4.46 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.45 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.44 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.35 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 3.95–4.07 (m, 1H (Ar-CH2-Ar) + 4H (-OCH2-)), 3.63–3.76 (m, 4H, -OCH2-), 3.16 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 3.15 (d, J = 13.4 Hz, 2H, Ar-CH2-Ar), 1.93–2.03 (m, 4H, -CH2-CH3), 1.82–1.92 (m, 4H, -CH2-CH3), 1.10 (t, J = 7.3 Hz, 6H, -CH3), 0.91 (t, J = 7.3 Hz, 3H, -CH3), 0.89 (t, J = 7.3 Hz, 6H, -CH3). 13C NMR (CDCl3, 125 MHz, 298 K) δ 160.47, 157.98, 157.88, 155.17, 155.16, 139.31, 137.99, 137.01, 136.89, 133.47, 133.22, 132.92, 132.67, 128.84, 128.78, 128.51, 127.57, 127.41, 127.33, 127.24, 126.21, 125.01, 122.10 (2C), 121.82, 121.78, 109.33, 101.42, 91.1, 88.83, 76.94, 76.90, 76.58, 76.44, 55.43 (2C), 31.04, 30.97, 30.96, 28.11, 23.50 (2C), 22.99, 22.97, 10.79, 10.78, 9.86, 9.84 ppm. HRMS (ESI+) calcd for C48H52O4 715.3757 [M + Na]+, found m/z 715.3764 [M + Na]+. IR (KBr) ν 2960, 2931, 2873, 1493, 1454, 1382, 1208, 1192, 1087, 1044, 965, 754, 689 cm−1.
Alkynylcalixarene 5c
Method A: Calixarene 3 (0.209 mmol, 150 mg), 4-ethynylanisole (0.230 mmol, 30 µL), CuI (0.0209 mmol, 4 mg) and Pd(PPh3)2Cl2 (0.011 mmol, 7 mg) were used under standard conditions; the product was purified by column chromatography on silica gel (DCM/cyclohexane 1:1) to afford 5c in 64% yield (97 mg) as a white solid.
Method B: Calixarene 3 (0.139 mmol, 106 mg), 4-ethynylanisole (0.556 mmol, 70 µL), CuI (0.028 mmol, 5 mg), piperidine (0.417 mmol, 40 µL) and Pd(PPh3)4 (0.0139 mmol, 16 mg) were used under standard conditions. The product 5c (58 mg, 58%) was obtained by column chromatography on silica gel (DCM/cyclohexane 50:50) as a white solid.
Data for compound 5c: M.p. = 146–149 °C. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.49 (d, 2H, J = 8.4 Hz, Ar-H) 7.22 (d, J = 7.7 Hz, 1H, Ar-H) 7.13 (d, J = 7.4 Hz, 2H, Ar-H) 7.10 (d, J = 7.8 Hz, 1H, Ar-H) 6.94 (t, J = 7.6 Hz, 1H, Ar-H) 6.88 (d, J = 8.4 Hz, 2H, Ar-H) 6.39 (d, J = 7.8 Hz, 1H, Ar-H), 6.29 (t, J = 7.6 Hz, 1H, Ar-H), 6.27 (t, J = 7.6 Hz, 1H, Ar-H), 6.18 (br d, J = 7.4 Hz, 2H, Ar-H), 6.17 (br d, J = 7.4 Hz, 1H, Ar-H), 4.50 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.49 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.48 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.38 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.12–4.02 (m, 4H, -OCH2-), 4.02 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 3.83 (s, 3H, -OCH3), 3.79–3.64 (m, 4H, -OCH2-), 3.19 (d, J = 13.4 Hz, 3H, Ar-CH2-Ar), 2.09–1.96 (m, 4H, -CH2-CH3), 1.96–1.84 (m, 4H, -CH2-CH3), 1.13 (t, J = 7.4 Hz, 6H, -CH3), 0.93 (t, J = 7.4 Hz, 3H, -CH3), 0.92 (t, J = 7.4 Hz, 3H, -CH3). 13C NMR (CDCl3, 125 MHz, 298 K) δ 160.35, 158.97, 158.99, 156.10 (2C), 140.02, 138.43, 137.93, 137.80, 135.25, 134.01, 133.89, 133.78 (2C), 133.58, 129.69, 129.62, 129.29, 128.35, 128.19, 128.11, 128.08, 126.76, 123.12, 122.92, 122.89, 122.58, 116.60, 114.64 (2C), 91.78, 88.45, 77.49, 77.46, 77.12, 76.99, 55.70, 31.34, 31.27 (2C), 28.30, 23.79 (2C), 23.27, 23.23, 11.03, 11.01, 10.06, 10.04 ppm. HRMS (ESI+) calcd for C49H54O5 745.3863 [M + Na]+, found m/z 775.3871 [M + Na]+. IR (KBr) ν 2960, 2932, 2920, 2874, 1511, 1454, 1249, 1208, 1192, 1105, 964, 830, 794, 775, 756 cm−1.
Alkynylcalixarene 5d
Method A: Calixarene 3 (0.209 mmol, 150 mg), 1-ethynyl-4-nitrobenzene (0.230 mmol, 34 mg), CuI (0.0209 mmol, 4 mg) and Pd(PPh3)2Cl2 (0.011 mmol, 7 mg) were used under standard conditions; the product was purified by column chromatography on silica gel (DCM/cyclohexane 1:1) to afford 5d in 27% yield (42 mg) as a yellow oil.
Method B: Calixarene 3 (0.097 mmol, 70 mg), 1-ethynyl-4-nitrobenzene (0.388 mmol, 62 mg), CuI (0.019 mmol, 4 mg), piperidine (0.291 mmol, 30 µL) and Pd(PPh3)4 (0.0097 mmol, 11 mg) were used under standard conditions. The product 5d (28 mg, 39%) was obtained by column chromatography on silica gel (DCM/cyclohexane 50:50) as a yellow oil.
Data for compound 5d: 1H NMR (CDCl3, 500 MHz, 298 K) δ 8.19 (d, J = 8.9 Hz, 2H, Ar-H), 7.63 (d, J = 8.9 Hz, 2H, Ar-H), 7.21 (d, J = 7.7 Hz, 1H, Ar-H), 7.08 (d, J = 7.7 Hz, 1H, Ar-H), 7.07 (d, J = 7.5 Hz, 1H, Ar-H), 6.89 (t, J = 7.5 Hz, 1H, Ar-H), 6.31 (dd, J = 7.7 Hz, J = 1.1 Hz, 1H, Ar-H), 6.26 (td, J = 7.5 Hz, J = 3.2 Hz, 2H, Ar-H), 6.19 (t, J = 6.2 Hz, 2H, Ar-H), 6.15 (d, J = 7.5 Hz, 1H, Ar-H), 4.49 (d, J = 13.3 Hz, 2H, Ar-CH2-Ar) 4.46 (dd, J = 13.3 Hz, J = 3.3 Hz 2H, Ar-CH2-Ar), 4.41 (d, J =13.3 Hz, 1H, Ar-CH2-Ar), 4.08–3.96 (m, 4H, -O-CH2-), 3.90 (d, 1H, J = 13.3 Hz, Ar-CH2-Ar), 3.78–3.65 (m, 4H, -O-CH2-), 3.19 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 3.16 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 3.73 (m, 2H, -O-CH2-), 1.99 (sextet, J = 7.7 Hz, 4H, -CH2-CH3), 1.89 (sextet, J = 7.7 Hz, 4H, -CH2-CH3), 1.11 (t, J = 7.5 Hz, 3H, -CH3), 1.10 (t, J = 7.5 Hz, 3H, -CH3), 0.92 (t, J = 7.5 Hz, 3H, -CH3), 0.91 (t, J = 7.5 Hz, 3H, -CH3).13C NMR (CDCl3, 125 MHz, 298 K) δ 158.02, 157.78, 155.24 (2C), 146.70, 139.56, 139.05, 136.87, 136.78, 133.62, 133.54, 132.59, 132.47, 132.10 (2C), 130.73, 128.80 (2C), 128.72, 127.71, 127.67, 127.21, 127.16, 126.46, 123.60 (2C), 122.11, 122.10, 121.85, 120.80, 95.05, 89.52, 76.97, 76.92, 76.68, 76.46, 31.10, 30.97 (2C), 28.20, 23.48, 23.47, 23.02, 22.98, 10.77, 10.76, 9.86, 9.85 ppm. HRMS (ESI+) calcd for C48H51NO6 760.3609 [M + Na]+, found m/z 760.3607 [M + Na]+. IR (KBr) ν 2960, 2929, 2874, 1589, 1454, 1382, 1340, 1208, 1192, 1087, 1004, 966, 756 cm−1.
Alkynylcalixarene 5e
Method A: Calixarene 3 (0.280 mmol, 205 mg), 3-ethynylpyridine (0.421 mmol, 40 µL), CuI (0.028 mmol, 5 mg) and Pd(PPh3)2Cl2 (0.014 mmol, 10 mg) were used under standard conditions; the product was purified by column chromatography on silica gel (DCM/cyclohexane 2:1) to afford 5e in 33% yield (64 mg) as a yellow oil.
Method B: Calixarene 3 (0.280 mmol, 204 mg), 3-ethynylpyridine (0.88 mmol, 90 µL), CuI (0.056 mmol, 11 mg), piperidine (0.841 mmol, 80 µL) and Pd(PPh3)4 (0.028 mmol, 32 mg) were used under standard conditions. The product 5e (91 mg, 47%) was obtained by column chromatography on silica gel (DCM/cyclohexane 2:1) as a yellow oil.
Data for compound 5e: 1H NMR (CDCl3, 500 MHz, 298 K) δ 8.61 (br s, 1H, Ar-H) 7.64 (td, J = 7.7 Hz, J = 1.5 Hz, 1H, Ar-H) 7.49 (d, J = 7.8 Hz, 1H, Ar-H) 7.27 (d, J = 7.8 Hz, 1H, Ar-H) 7.20 (dd, J = 7.8 Hz, J = 4.8 Hz, 1H, Ar-H) 7.08 (d, J = 7.4 Hz, 3H, Ar-H) 6.89 (t, J = 7.4 Hz, 1H, Ar-H), 6.33 (d, J = 7.3 Hz, 1H, Ar-H), 6.21–6.28 (m, 2H, Ar-H), 6.10–6.19 (m, 3H, Ar-H), 4.48 (d, J = 13.0 Hz, 1H, Ar-CH2-Ar), 4.46 (d, J = 13.0 Hz, 1H, Ar-CH2-Ar), 4.45 (d, J = 13.0 Hz, 1H, Ar-CH2-Ar), 4.38 (d, J = 13.0 Hz, 1H, Ar-CH2-Ar), 3.94–4.10 (m, 1H (Ar-CH2-Ar) + 4H (-OCH2-)), 3.62–3.79 (m, 4H, -OCH2-), 3.18 (d, 1H, J = 13.0 Hz, 1H, Ar-CH2-Ar), 3.15 (d, 1H, J = 13.0 Hz, 2H, Ar-CH2-Ar), 1.93–2.03 (m, 4H, -CH2-CH3), 1.84–1.92 (m, 4H, -CH2-CH3), 1.10 (t, J = 7.3 Hz, 6H, -CH3), 0.91 (t, J = 7.3 Hz, 6H, -CH3), 0.90 (t, J = 7.3 Hz, 6H, -CH3).13C NMR (CDCl3, 125 MHz, 298 K) δ 157.98, 157.89, 155.19, 150.02, 143.88, 139.80, 138.70, 137.02, 136.90, 136.04, 133.50, 133.24, 132.79, 132.57, 128.86, 128.80, 128.61, 127.62, 127.45, 127.38, 127.26, 126.70, 122.46, 122.14, 122.12, 121.80, 121.04, 90.56, 89.46, 76.97, 76.90, 76.64, 76.47, 31.10, 31.00, 30.98, 23.51 (2C), 23.01, 23.99, 10.81, 10.79, 9.87, 9.86 ppm. HRMS (ESI+) calcd for C47H51NO4 716.3710 [M + Na]+, found m/z 716.3710 [M + Na]+. IR (KBr) ν 2960, 2931, 2873, 1580, 1455, 1383, 1209, 1192, 1087, 1066, 1005, 956, 756 cm−1.
Alkynylcalixarene 5f
Method A: Calixarene 3 (0.349 mmol, 250 mg), trimethylsilylacetylene (0.418 mmol, 60 µL), CuI (0.035 mmol, 7 mg) and Pd(PPh3)2Cl2 (0.017 mmol, 12 mg) were used under standard conditions; the product was purified by column chromatography on silica gel (DCM/cyclohexane 1:1) to afford 5f in 83% yield (199 mg) as a yellow oil.
Method B: Calixarene 3 (0.138 mmol, 104 mg), trimethylsilylacetylene (0.552 mmol, 78 µL), CuI (0.028 mmol, 5 mg), piperidine (0.417 mmol, 40 µL) and Pd(PPh3)4 (0.014 mmol, 16 mg) were used under standard conditions. The product 5f (71 mg, 75%) was obtained by column chromatography on silica gel (DCM/cyclohexane 1:1) as a yellow oil.
Data for compound 5f: 1H NMR (CDCl3, 500 MHz, 298 K) δ 7.14 (d, 1H, J = 7.8 Hz, Ar-H) 7.10 (m, 2H, Ar-H) 7.03 (d, J = 7.8 Hz, 2H, Ar-H) 6.91 (t, J = 7.4 Hz, 1H, Ar-H) 6.27 (dd, J = 7.7 Hz, J = 1.7 Hz, 1H, Ar-H) 6.25 (t, J = 7.4 Hz, 1H, Ar-H) 6.20 (t, J = 7.4 Hz, 1H, Ar-H), 6.15–6.10 (m, 2H, Ar-H), 6.07 (dd, J = 7.4 Hz, J = 1.7 Hz, 1H, Ar-H), 4.43 (d, J = 13.5 Hz, 1H, Ar-CH2-Ar), 4.44 (d, J = 13.5 Hz, 2H, Ar-CH2-Ar), 4.28 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.04–3.95 (m, 4H, -OCH2), 4.48 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.38 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.12–4.02 (m, 4H, -OCH2-), 3.90 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 3.76–3.60 (m, 4H, -OCH2-), 3.14 (d, J = 13.4 Hz, 2H, Ar-CH2-Ar), 3.13 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 2.02–1.90 (m, 4H, -CH2-CH3), 1.91–1.80 (m, 4H, -CH2-CH3), 1.09 (t, J = 7.4 Hz, 3H, -CH3), 1.08 (t, J = 7.4 Hz, 3H, -CH3), 0.87 (t, J = 7.4 Hz, 6H, -CH3), 0.21 (s, 9H, -CH3) 13C NMR (CDCl3, 100 MHz, 298 K) δ 158.40, 158.36, 155.55, 155.50, 140.07, 138.56, 137.43, 137.35, 133.70, 133.47, 133.28, 132.93, 129.22, 129.12, 128.71, 127.84, 127.65, 127.54, 127.50, 126.79, 122.41, 122.36, 122.20, 122.06, 105.20, 96.13, 76.98 (2C), 76.57, 76.49, 30.82, 30.77, 30.74, 27.64, 23.28 (2C), 22.71 (2C), 10.52 (2C), 9.51 (2C), 0.28 (3C).ppm. HRMS (ESI+) calcd for C45H56SiO4 711.3840 [M + Na]+, found m/z 711.3847 [M + Na]+. IR (KBr) ν 2960, 2932, 2874, 1455, 1383, 1247, 1207, 1192, 1088, 1006, 966, 841, 756 cm−1.
3.2.2. Preparation of Bridged Calixarene 7b
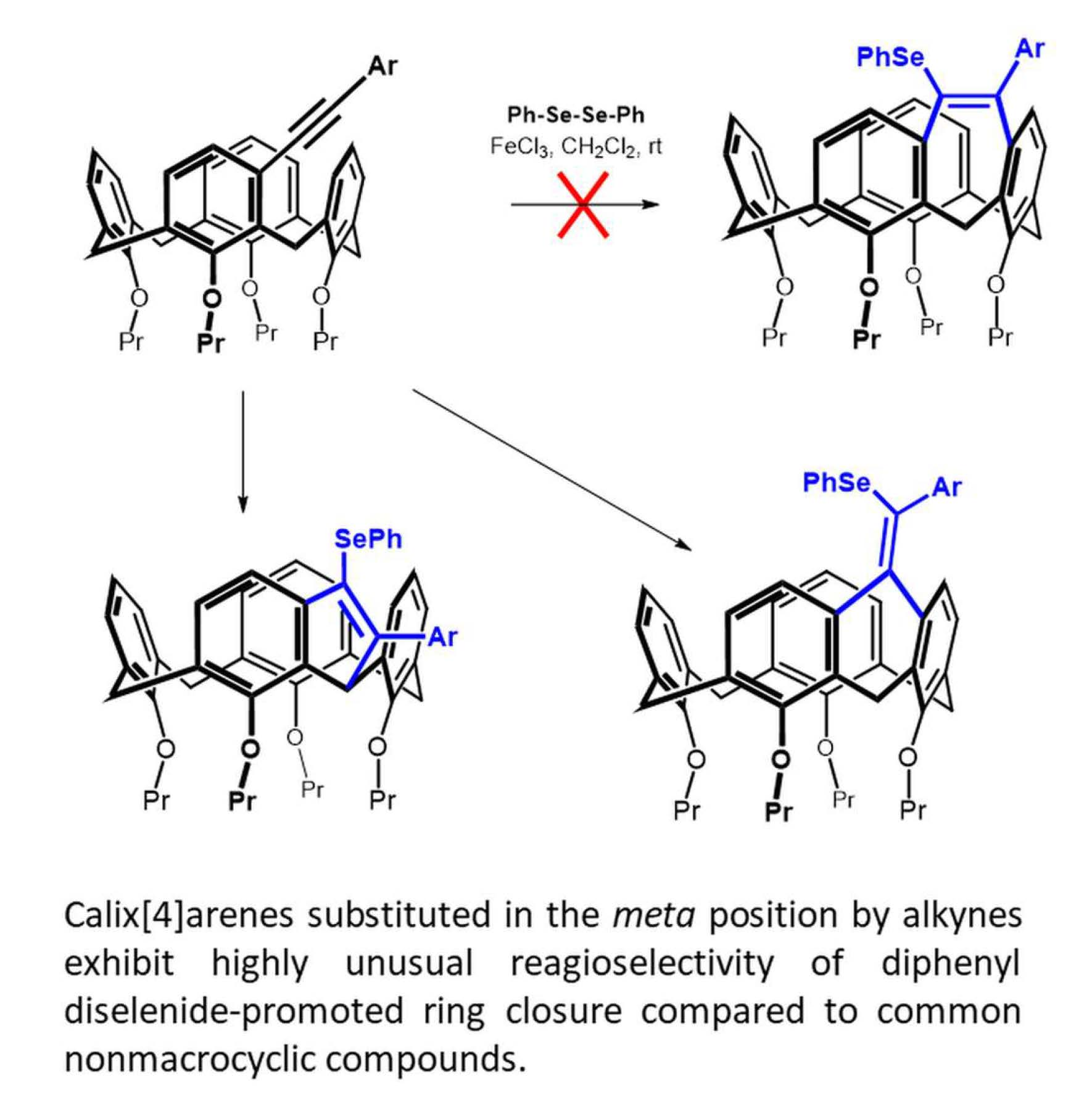
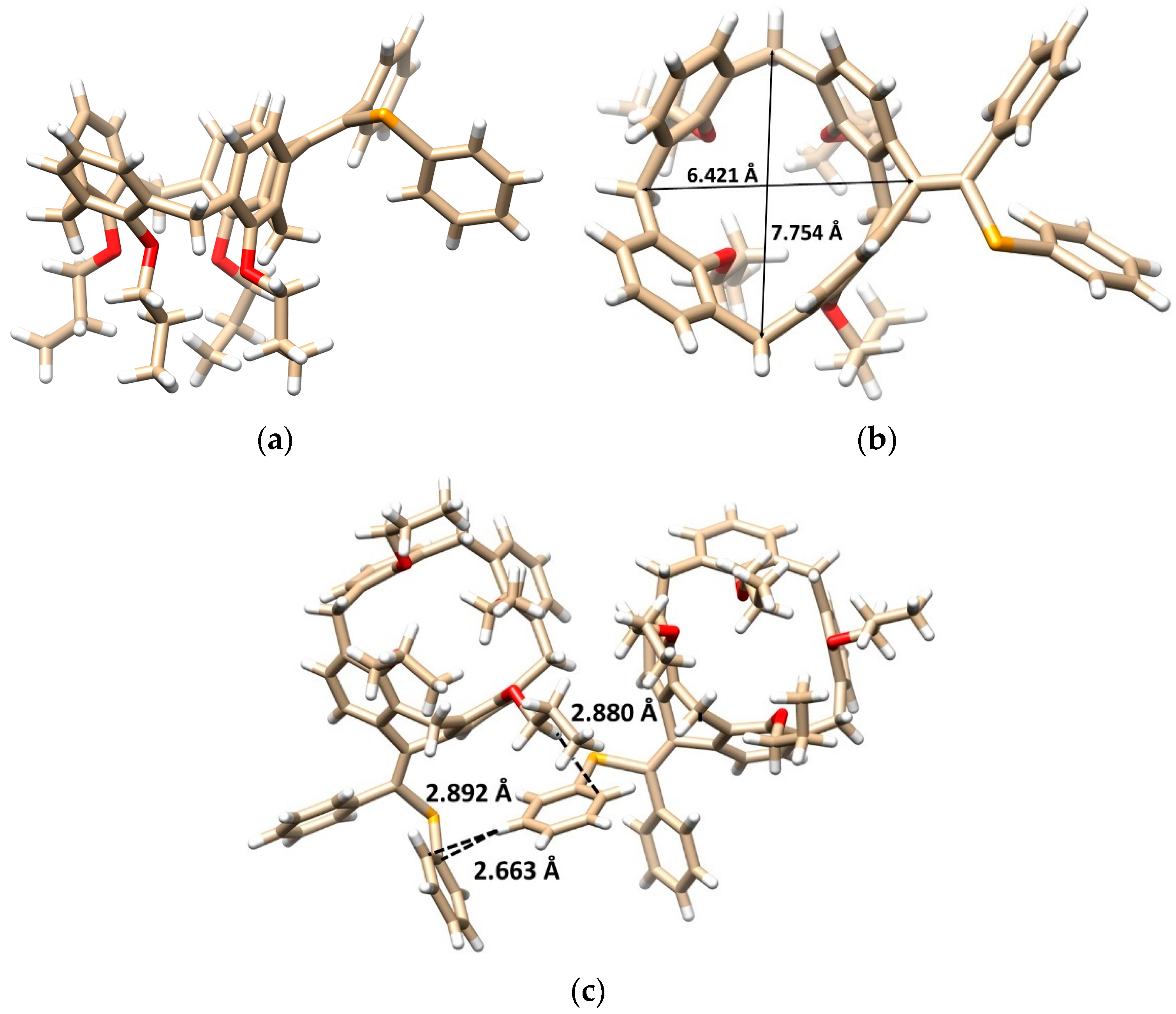
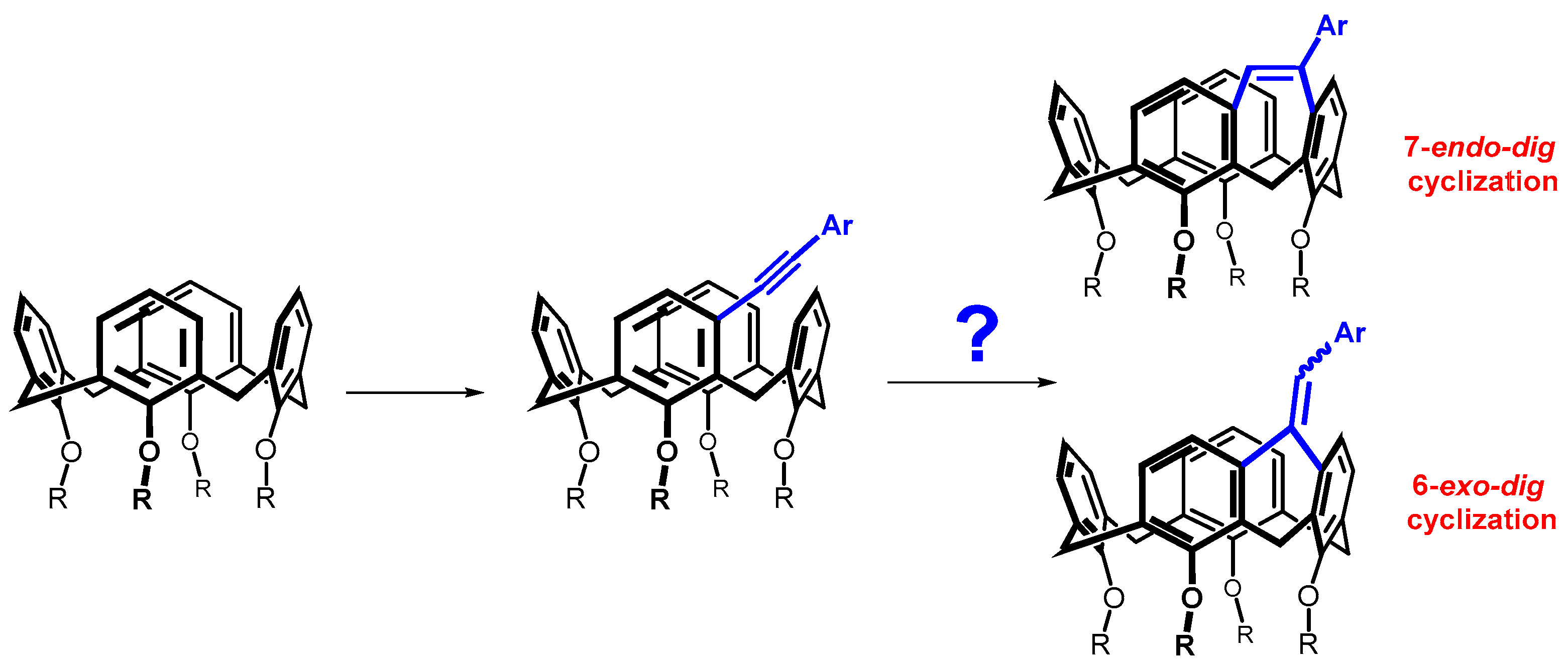
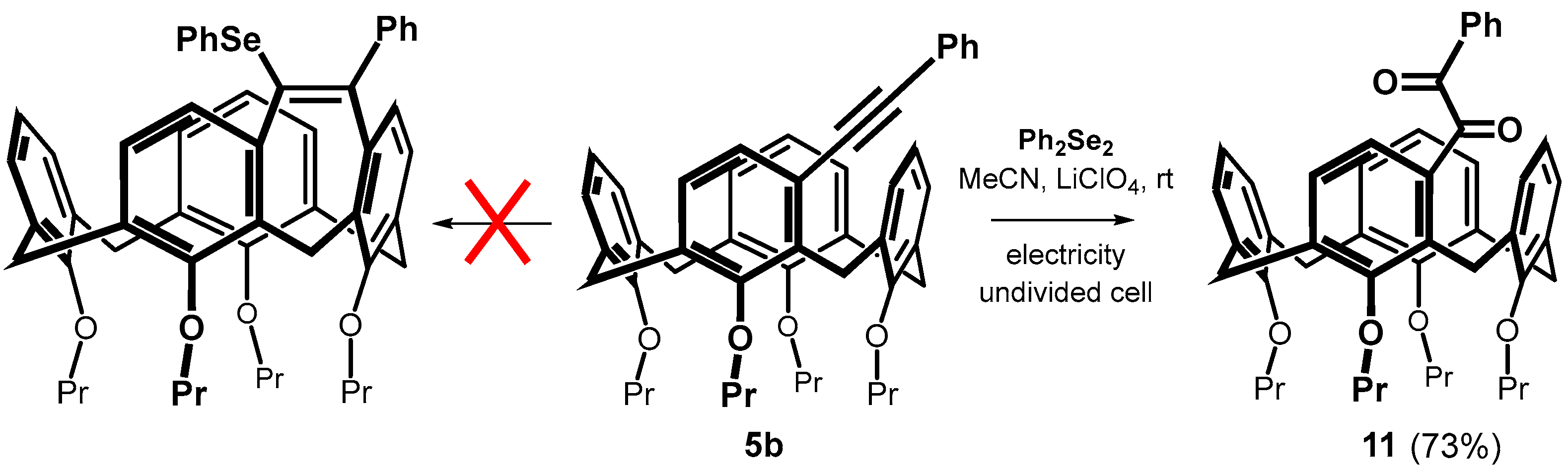
Diphenyl diselenide (0.609 mmol, 190 mg) and FeCl3 (0.752 mmol, 122 mg) were dissolved in 4 mL of dry dichloromethane in a Schlenk flask under an argon atmosphere. The resulting solution was stirred for 15 min at room temperature. After this time, the solution changed color to brown-red. Calixarene 5b (0.301 mmol, 209 mg) dissolved in 2 mL of dry dichloromethane was added to the above solution, and the resulting mixture was heated at 30 °C for 4 h. Dichloromethane (20 mL) was then added to the reaction mixture, the organic layer was washed with a saturated solution of NH4Cl, dried with MgSO4 and concentrated under vacuum. The residue was purified by column chromatography on silica gel (DCM/cyclohexane 1:1) to provide the respective product 7b in 20% yield (51 mg) as a yellow solid.
Data for compound 7b: M.p. = 117–120 °C. 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.24 (dd, J = 8.0 Hz, J = 1.8 Hz, 2H, Ar-H), 7.19 (dd, J = 8.0 Hz, J = 1.8 Hz, 2H, Ar-H), 7.03–6.95 (m, 7H, Ar-H) 6.93 (d, J = 7.3 Hz, 1H, Ar-H), 6.89 (d, J = 8.0 Hz, 1H, Ar-H), 6.87 (dd, J = 8.0 Hz, J = 1.5 Hz, 1H, Ar-H), 6.82 (dd, J = 7.8 Hz, J = 1.5 Hz, 1H, Ar-H), 6.75 (d, J = 9.0 Hz, 1H, Ar-H), 6.73 (t, J = 7.6 Hz, 1H, Ar-H), 6.61 (t, J = 7.4 Hz, 1H, Ar-H), 6.43 (d, J = 7.8 Hz, 1H, Ar-H), 5.98 (d, J = 7.8 Hz, 1H, Ar-H), 4.70 (d, J = 13.4 Hz, 1H, Ar-CH2-Ar), 4.50 (d, J = 12.5 Hz, 1H, Ar-CH2-Ar), 4.41 (d, J = 12.0 Hz, 1H, Ar-CH2-Ar), 4.33 (d, J = 12.5 Hz, 1H, Ar-CH2-Ar), 4.04–3.79 (m, 6H, -OCH2-), 3.64–3.54 (m, 2H, -OCH2-), 3.28 (d, J = 13.2 Hz, 1H, Ar-CH2-Ar), 3.27 (d, J = 12.5 Hz, 1H, Ar-CH2-Ar), 3.11 (d, J = 12.5 Hz, 1H, Ar-CH2-Ar), 3.07 (d, J = 12.5 Hz, 1H, Ar-CH2-Ar), 2.21 (sextet, J = 7.7 Hz, 2H, -CH2-CH3), 2.13–2.01 (m, 2H, -CH2-CH3), 2.01–1.83 (m, 4H, -CH2-CH3), 1.20 (t, J = 7.4 Hz, 3H, -CH3), 1.19 (t, J = 7.4 Hz, 3H, -CH3), 1.00 (t, J = 7.4 Hz, 3H, -CH3), 0.99 (t, J = 7.4 Hz, 3H, -CH3). 13C NMR (CDCl3, 100 MHz, 298 K) δ 156.32, 155.80, 154.80, 154.77, 140.82, 140.37, 140.36, 140.08, 136.54, 136.23, 135.04, 134.42, 134.18, 132.98, 132.71, 132.70, 132.21, 131.16, 130.97 (2C), 130.39, 129.27, 128.86, 128.41 (2C), 128.09, 127.40, 127.37 (2C), 126.89, 126.58, 126.48, 126.44, 123.27, 123.05, 123.01, 122.94, 77.48, 77.45, 76.99, 76.70, 31.79, 31.56, 29.19, 24.82, 23.77, 23.73, 23.12, 22.99, 11.10 (2C), 10.29, 10.15 ppm. HRMS (ESI+) calcd for C54H56SeO4 871.3236 [M + Na]+, found m/z 871.3246 [M + Na]+. IR (KBr) ν 2959, 2924, 2872, 1455, 1250, 1208, 1066, 1007, 960, 734, 690 cm−1.
3.2.3. Preparation of Bridged Calixarene 8b
Diphenyl diselenide (0.28 mmol, 90 mg) and FeCl3 (0.351 mmol, 57 mg) were dissolved in 3 mL of dry DCE in a Schlenk flask under an argon atmosphere, and the resulting solution was stirred for 15 min at room temperature. After this time, the color of the solution turned to brown-red. Calixarene 5b (0.144 mmol, 100 mg) dissolved in 1 mL of dry DCE was added, and the reaction mixture was heated to 85 °C for 4 h. The eluent was then evaporated under reduced pressure, dichloromethane (20 mL) was added to the crude product, washed with a saturated solution of NH4Cl, dried with MgSO4 and then concentrated under vacuum. The residue was purified by preparative thin layer chromatography on silica gel (DCM/cyclohexane 1:1) to provide the methylene bridged product 8b in 10% yield (12 mg) as a white solid.
Data for compound 8b: M.p. = 101–104 °C. 1H NMR (CDCl3, 400 MHz, 298 K) δ 8.38 (s, 1H, -OH), 8.25 (s, 1H, -OH), 7.45 (d, J = 7.9 Hz, 2H, Ar-H), 7.39 (d, J = 1.4 Hz, 1H, Ar-H), 7.34 (t, J = 7.0 Hz, 3H, Ar-H), 7.30–7.11 (m, 10H, Ar-H) 7.07 (d, J = 7.3 Hz, 1H, Ar-H), 6.87 (d, J = 7.3 Hz, 1H, Ar-H), 6.83 (d, J = 7.3 Hz, 2H, Ar-H), 6.74 (d, J = 7.5 Hz, 1H, Ar-H), 6.73 (t, J = 7.4 Hz, 1H, Ar-H), 6.59 (t, J = 7.7 Hz, 1H, Ar-H), 6.50 (d, J = 7.2 Hz, 1H, Ar-H), 5.49 (s, 1H, Ar-CH(R)-Ar), 4.51 (d, J = 12.4 Hz, 1H, Ar-CH2-Ar), 4.43 (d, J = 12.5 Hz, 1H, Ar-CH2-Ar), 4.18–4.04 (m, 2H, -OCH2-), 4.03–3.90 (m, 2H, -OCH2-), 3.43 (d, J = 13.0 Hz, 1H, Ar-CH2-Ar), 3.35 (d, J = 13.0 Hz, 1H, Ar-CH2-Ar), 3.26 (d, J = 12.4 Hz, 1H, Ar-CH2-Ar), 2.19–1.96 (m, 4H, -CH2-CH3), 1.37 (t, J = 7.3 Hz, 3H, -CH3), 1.30 (t, J = 7.4 Hz, 3H, -CH3). 13C NMR (CDCl3, 125 MHz, 298 K) δ 154.14, 153.59, 153.13, 152.32, 151.24, 144.49, 135.99, 134.72, 134.00, 133.82, 132.04, 131.70, 130.81, 130.35, 130.24 (2C), 130.24, 130.06, 130.02, 129.72 (2C), 129.46 (2C), 129.32 (2C), 129.21, 129.17, 128.68, 128.56, 128.23 (2C), 128.01, 127.57, 126.25, 125.94, 125.76, 125.50, 125.22, 116.72, 113.61, 78.29, 78.21, 50.51, 32.11, 31.45, 30.09, 23.88, 23.69, 11.18, 11.06 ppm. HRMS (ESI+) calcd for C54H48Se2O4 943.1775 [M + Na]+, found m/z 943.1785 [M + Na]+.
3.2.4. Preparation of Bridged Calixarenes 9c and 10c
Diphenyl diselenide (0.170 mmol, 53 mg) and FeCl3 (0.213 mmol, 34 mg) were dissolved in 3 mL of dry dichloromethane in a Schlenk flask under an argon atmosphere. The resulting solution was stirred for 15 min at room temperature. After this time, the solution changed color to brown-red. Calixarene 5c (0.065 mmol, 85 mg) dissolved in 1 mL of dry dichloromethane was added, and the reaction mixture was heated at 30 °C for 4 h. Dichloromethane (20 mL) was added to the mixture, the organic layer was washed with a saturated solution of NH4Cl, dried over MgSO4, and concentrated under vacuum. The residue was purified by column chromatography on a silica gel (DCM/cyclohexane 1:1) to provide the respective products 9c in 37% yield (27 mg) and 10c in 15% yield (11 mg) as yellow oils.
Data for compound 9c: 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.41 (dd, J = 7.4 Hz, J = 1.5 Hz, 2H, Ar-H), 7.31 (d, J = 8.4 Hz, 2H, Ar-H), 7.23–7.15 (m, 4H, Ar-H) 7.13 (dd, J = 7.5 Hz, J = 1.4 Hz, 1H, Ar-H), 7.07 (d, J = 7.5 Hz, 1H, Ar-H), 6.95 (t, J = 7.3 Hz, 1H, Ar-H), 6.87 (d, J = 7.4 Hz, 1H, Ar-H), 6.77 (d, J = 8.9 Hz, 2H, Ar-H), 6.20 (t, J = 7.4 Hz, 1H, Ar-H), 6.14–6.07 (m, 3H, Ar-H), 6.02 (dd, J = 7.5 Hz, J = 1.3 Hz, 1H, Ar-H), 5.80 (dd, J = 7.5 Hz, J = 1.5 Hz, 1H, Ar-H), 5.70 (s, 1H, Ar-CH(R)-Ar), 4.49 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 4.44 (d, J = 13.3 Hz, 2H, Ar-CH2-Ar), 4.34 (td, J = 11.0 Hz, J = 5.3 Hz, 1H, -OCH2-), 4.23–4.10 (m, 2H, -OCH2-), 4.04–3.94 (m, 1H, -OCH2-), 3.74 (s, 3H, -OCH3), 3.71–3.64 (m, 3H, -OCH2-), 3.54–3.46 (m, 1H, -OCH2-), 3.19 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 3.15 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 3.13 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 2.04–1.81 (m, 8H, -CH2-CH3), 1.14 (t, J = 7.5 Hz, 3H, -CH3), 1.10 (t, J = 7.4 Hz, 3H, -CH3), 0.91 (t, J = 7.5 Hz, 3H, -CH3), 0.89 (t, J = 7.4 Hz, 3H, -CH3). We cannot acquire reliable 13C NMR spectra due to degradation of compound. HRMS (ESI+) calcd for C55H58SeO5 901.3341 [M + Na]+, found m/z 901.3345 [M + Na]+.
Data for compound 10c: 1H NMR (CDCl3, 400 MHz, 298 K) δ 7.40 (dd, J = 7.4 Hz, J = 1.5 Hz, 2H, Ar-H), 7.37 (d, J = 8.6 Hz, 2H, Ar-H), 7.22–7.14 (m, 3H, Ar-H), 7.10 (d, J = 7.6 Hz, 3H, Ar-H), 6.93 (d, J = 7.6 Hz, 1H, Ar-H), 6.81 (d, J = 7.6 Hz, 1H, Ar-H), 6.77 (d, J = 8.6 Hz, 2H, Ar-H), 6.46–6.34 (m, 3H, Ar-H), 6.27 (~dd, J = 7.6 Hz, J = 1.5 Hz, 1H, Ar-H), 6.22 (t, J = 7.6 Hz, 1H, Ar-H), 5.98 (dd, J = 7.6 Hz, J = 1.5 Hz, 1H, Ar-H), 5.74 (s, 1H, Ar-CH(R)-Ar), 4.42 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 4.40 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 4.37 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 4.28–4.19 (m, 1H, -OCH2-), 4.00–3.92 (m, 1H, -OCH2-), 3.77–3.72 (m, 3H + 3H, -OCH2-, -OCH3), 3.65–3.59 (m, 1H, -OCH2-), 3.33 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 3.23 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 3.22 (d, J = 13.3 Hz, 1H, Ar-CH2-Ar), 2.26–2.20 (m, 1H, -CH2-CH3), 2.15–2.07 (m, 1H, -CH2-CH3), 1.97–1.84 (m, 4H, -CH2-CH3), 1.15 (t, J = 7.3 Hz, 3H, -CH3), 1.10 (t, J = 7.3 Hz, 3H, -CH3), 0.93 (t, J = 7.4 Hz, 3H, -CH3). We cannot acquire 13C NMR spectra due to degradation of compound. HRMS (ESI+) calcd for C52H52SeO5 859.2881 [M + Na]+, found m/z 859.2881 [M + Na]+.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}