
In Silico Identification and Validation of Organic Triazole Based Ligands as Potential Inhibitory Drug Compounds of SARS-CoV-2 Main Protease



Abstract
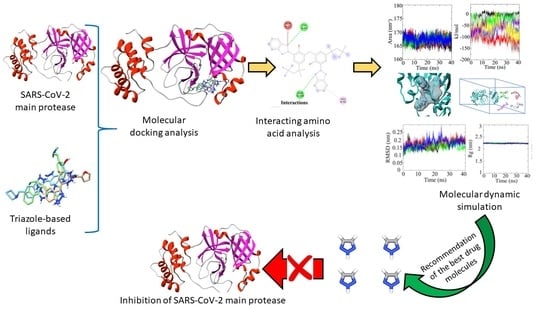
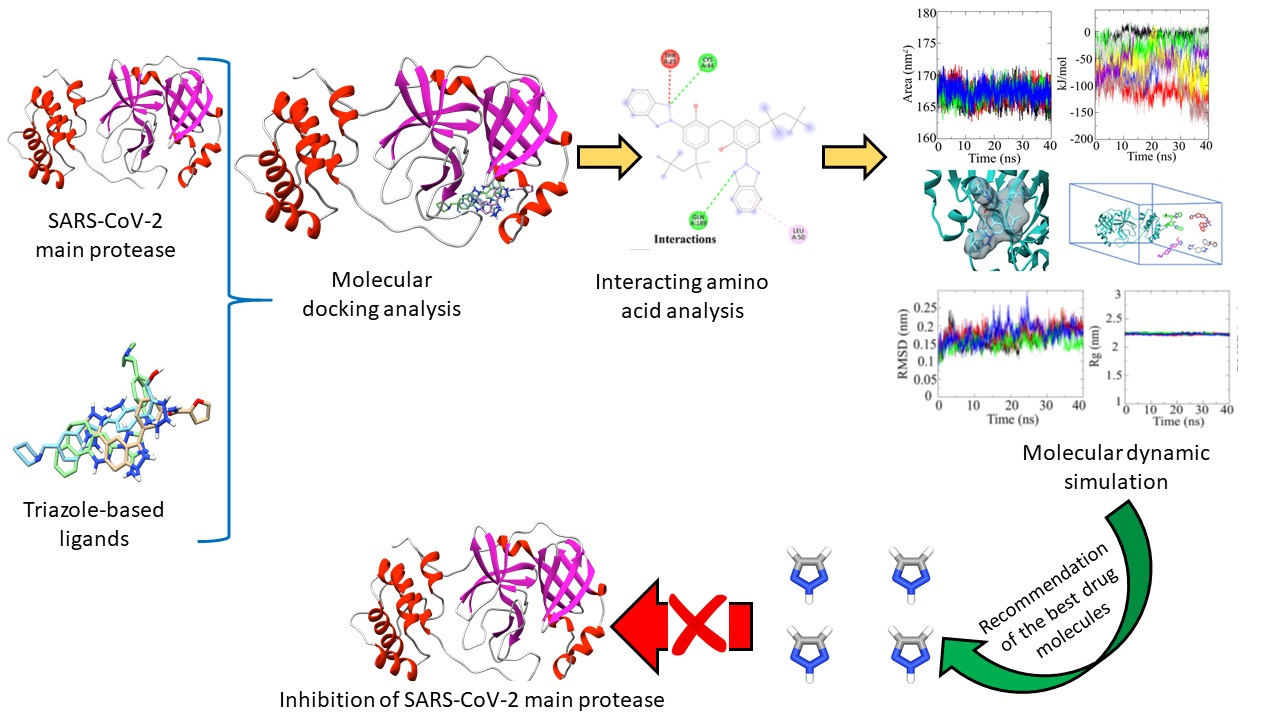
:
1. Introduction
2. Results
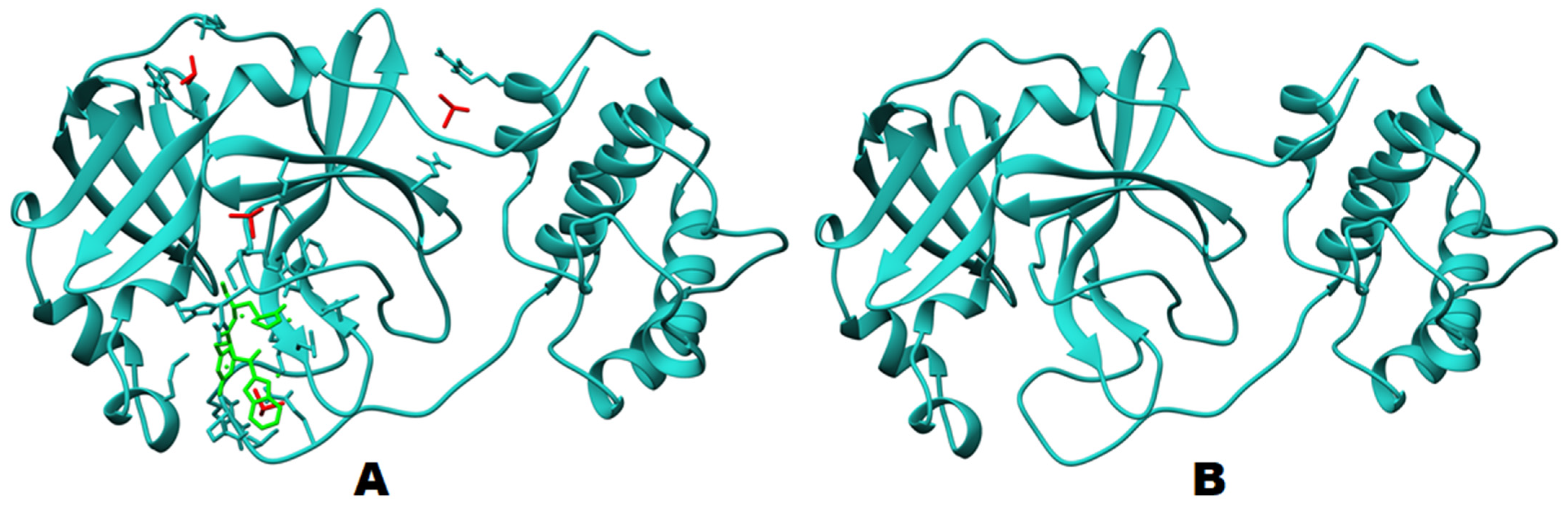
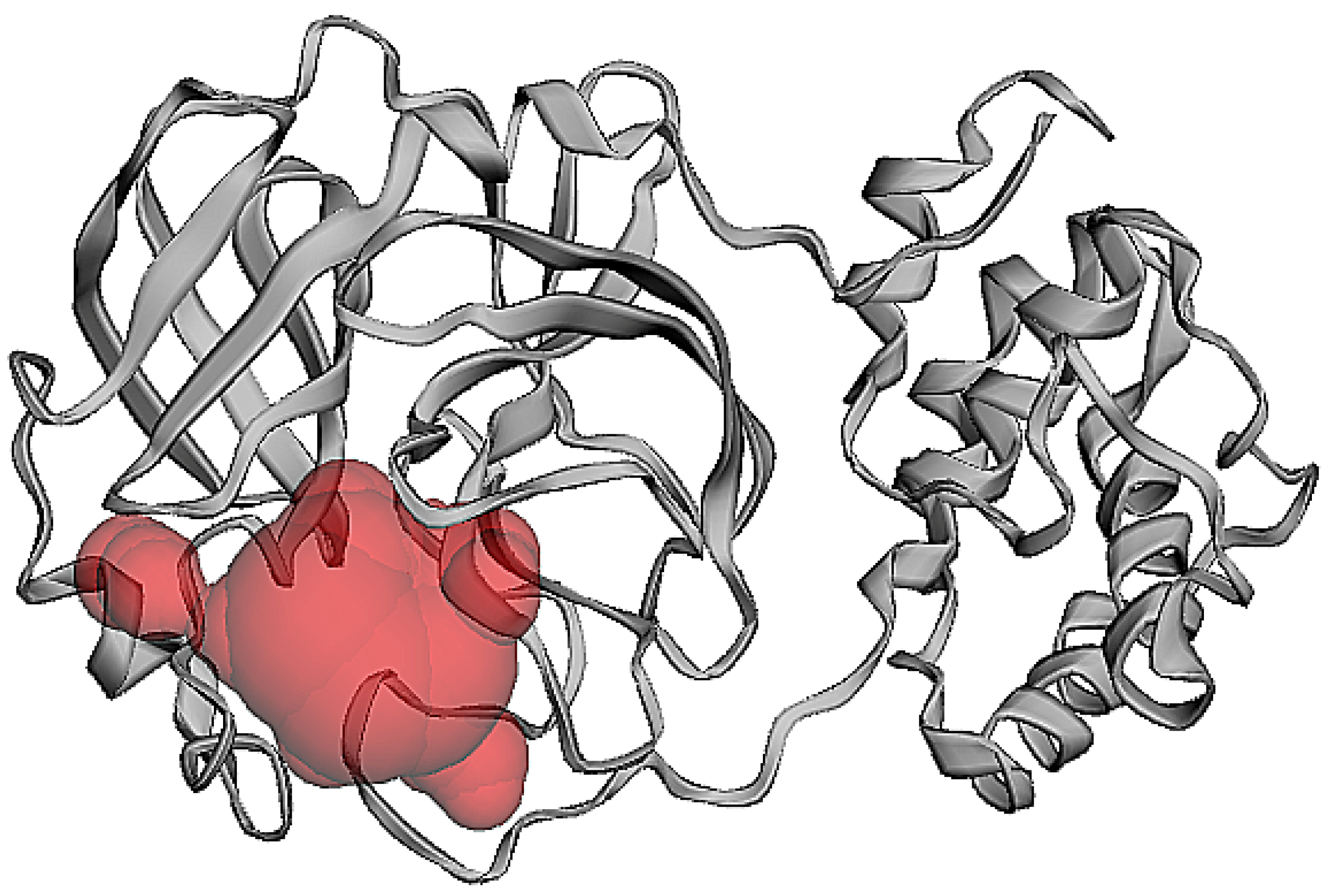
2.1. Structural Analysis
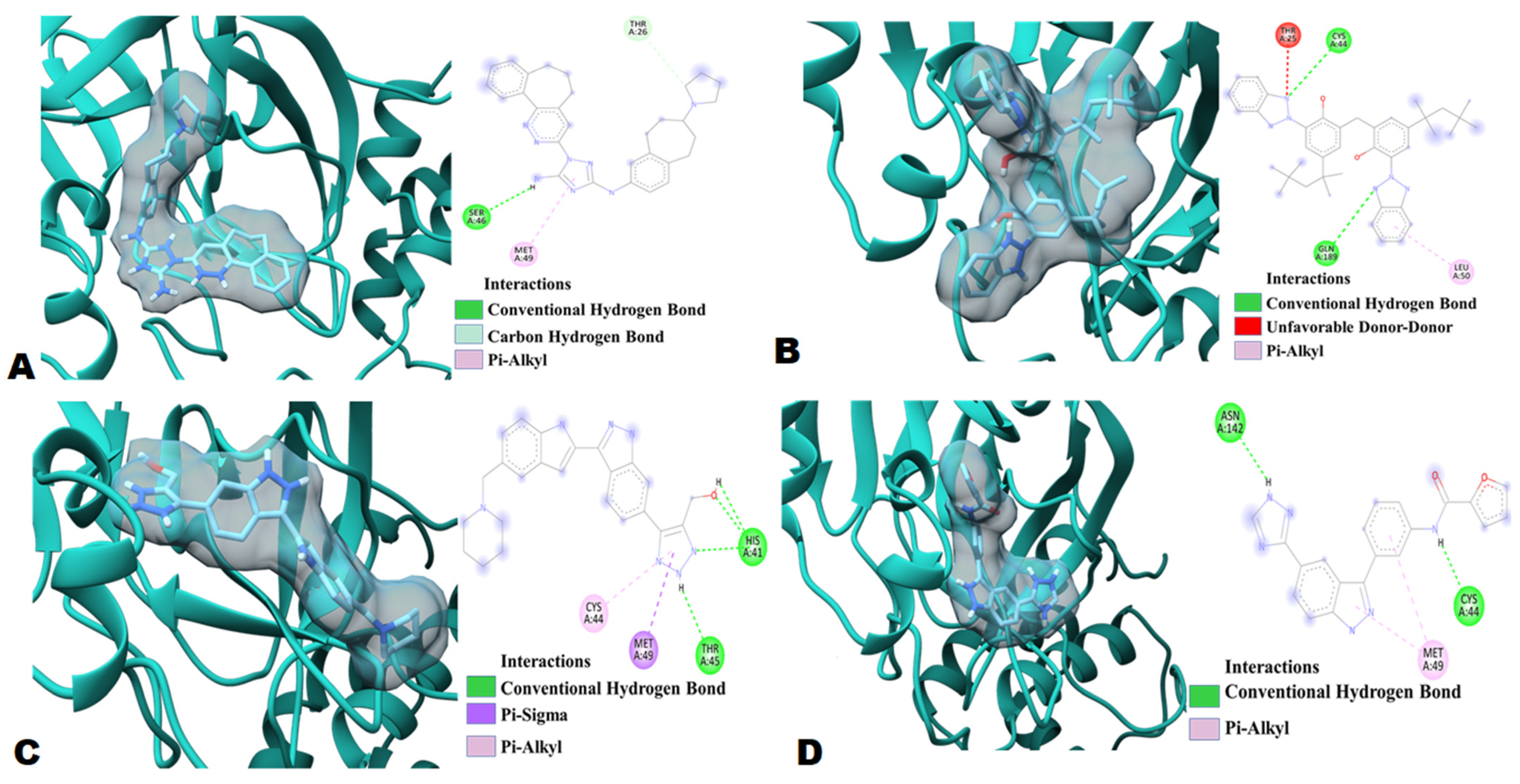
2.2. Molecular Docking
2.3. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Analysis
2.3.1. Absorption
2.3.2. Distribution
2.3.3. Metabolism
2.3.4. Excretion
2.3.5. Toxicity
2.4. In Silico Antiviral Prediction
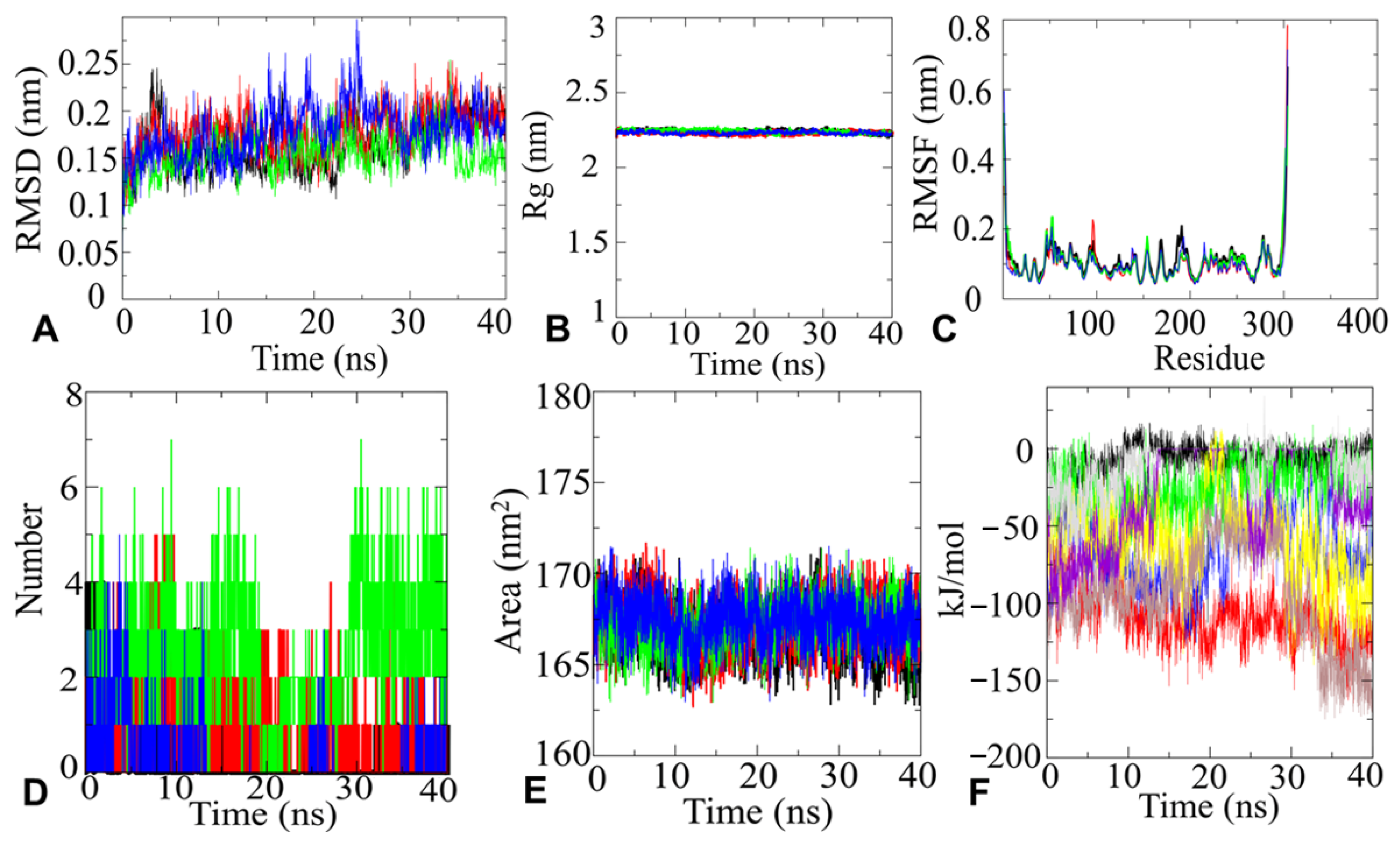
2.4.1. MD Simulation and Analysis
2.4.2. RMSD Analysis
2.4.3. Rg Analysis
2.4.4. RMSF Analysis
2.4.5. H-Bonds Analysis
2.4.6. SASA Analysis
2.4.7. Interaction Energy Analysis
3. Materials and Methods
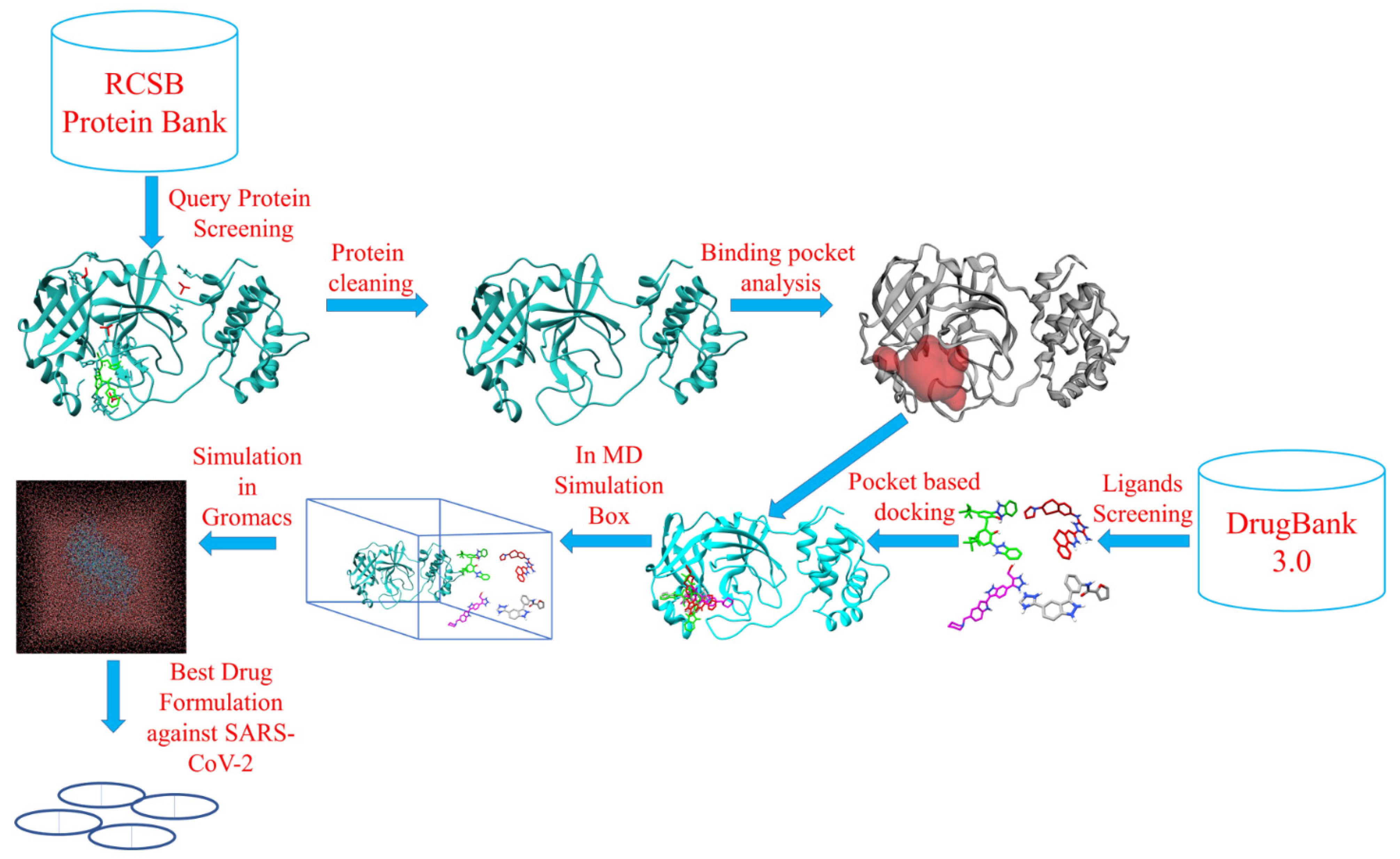
3.1. Target and Ligand Preparation
3.2. Protein Pocket Analysis
3.3. Molecular Docking and Interaction Analysis
3.4. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Analysis
3.5. In Silico Antiviral Assay
3.6. MD Simulation Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Kumar, S.; Nyodu, R.; Maurya, V.K.; Saxena, S.K. Morphology, Genome Organization, Replication, and Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). In Coronavirus Disease 2019 (COVID-19); Springer: Berlin, Germany, 2020; pp. 23–31. [Google Scholar] [CrossRef]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: New York, NY, USA, 2015; pp. 1–23. [Google Scholar] [CrossRef] [Green Version]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef] [PubMed]
- Trezza, A.; Iovinelli, D.; Santucci, A.; Prischi, F.; Spiga, O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 13866. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cava, C.; Bertoli, G.; Castiglioni, I. In Silico Discovery of Candidate Drugs against COVID-19. Viruses 2020, 12, 404. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Liang, J.; Pitsillou, E.; Karagiannis, C.; Darmawan, K.K.; Ng, K.; Hung, A.; Karagiannis, T.C. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: Molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020, 87, 107292. [Google Scholar] [CrossRef]
- Gaudêncio, S.P.; Pereira, F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2020, 18, 633. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, X.; Liu, H.; Liu, S.; Yu, H.; Wang, X.; Qin, Y.; Li, P. Preparation of New Sargassum fusiforme Polysaccharide Long-Chain Alkyl Group Nanomicelles and Their Antiviral Properties against ALV-J. Molecules 2021, 26, 3265. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zhang, Y. Colloidal nanoparticle inks for printing functional devices: Emerging trends and future prospects. J. Mater. Chem. A 2019, 7, 23301–23336. [Google Scholar] [CrossRef]
- Zeng, M.; Chen, M.; Huang, D.; Lei, S.; Zhang, X.; Wang, L.; Cheng, Z. Engineered two-dimensional nanomaterials: An emerging paradigm for water purification and monitoring. Mater. Horiz. 2021, 8, 758–802. [Google Scholar] [CrossRef]
- Witika, B.A.; Makoni, P.A.; Matafwali, S.K.; Mweetwa, L.L.; Shandele, G.C.; Walker, R.B. Enhancement of Biological and Pharmacological Properties of an Encapsulated Polyphenol: Curcumin. Molecules 2021, 26, 4244. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Gomes, D.; Esteruelas, G.; Bonilla, L.; Lopez-Machado, A.L.; Galindo, R.; Cano, A.; Espina, M.; Ettcheto, M.; Camins, A.; et al. Metal-Based Nanoparticles as Antimicrobial Agents: An Overview. Nanomaterials 2020, 10, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, V.P.; Mazumdar, A.; Kopel, P.; Mukherjee, S.; Vítek, P.; Michalkova, H.; Vaculovičová, M.; Moulick, A. A Novel Ruthenium Based Coordination Compound Against Pathogenic Bacteria. Int. J. Mol. Sci. 2020, 21, 2656. [Google Scholar] [CrossRef] [PubMed]
- Murcia, R.A.; Leal, S.M.; Roa, M.V.; Nagles, E.; Muñoz-Castro, A.; Hurtado, J.J. Development of Antibacterial and Antifungal Triazole Chromium(III) and Cobalt(II) Complexes: Synthesis and Biological Activity Evaluations. Molecules 2018, 23, 2013. [Google Scholar] [CrossRef] [Green Version]
- Sumrra, S.H.; Kausar, S.; Raza, M.A.; Zubair, M.; Zafar, M.N.; Nadeem, M.A.; Mughal, E.U.; Chohan, Z.H.; Mushtaq, F.; Rashid, U. Metal based triazole compounds: Their synthesis, computational, antioxidant, enzyme inhibition and antimicrobial properties. J. Mol. Struct. 2018, 1168, 202–211. [Google Scholar] [CrossRef]
- Karypidou, K.; Ribone, S.R.; Quevedo, M.A.; Persoons, L.; Pannecouque, C.; Helsen, C.; Claessens, F.; Dehaen, W. Synthesis, biological evaluation and molecular modeling of a novel series of fused 1,2,3-triazoles as potential anti-coronavirus agents. Bioorganic Med. Chem. Lett. 2018, 28, 3472–3476. [Google Scholar] [CrossRef]
- Seck, I.; Nguemo, F. Triazole, imidazole, and thiazole-based compounds as potential agents against coronavirus. Results Chem. 2021, 3, 100132. [Google Scholar] [CrossRef]
- Kumari, A.; Rajput, V.S.; Nagpal, P.; Kukrety, H.; Grover, S.; Grover, A. Dual inhibition of SARS-CoV-2 spike and main protease through a repurposed drug, rutin. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Chaudhary, M.; Qamar, I.; Singh, N.; Nain, V. In silico identification and validation of natural antiviral compounds as potential inhibitors of SARS-CoV-2 methyltransferase. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef]
- Saha, S.; Nandi, R.; Vishwakarma, P.; Prakash, A.; Kumar, D. Discovering Potential RNA Dependent RNA Polymerase Inhibitors as Prospective Drugs Against COVID-19: An in silico Approach. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Tamura, S.; Yang, G.-M.; Yasueda, N.; Matsuura, Y.; Komoda, Y.; Murakami, N. Tellimagrandin I, HCV invasion inhibitor from Rosae Rugosae Flos. Bioorganic Med. Chem. Lett. 2010, 20, 1598–1600. [Google Scholar] [CrossRef]
- Bhowmik, D.; Nandi, R.; Jagadeesan, R.; Kumar, N.; Prakash, A.; Kumar, D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect. Genet. Evol. 2020, 84, 104451. [Google Scholar] [CrossRef]
- Sinha, M.; Jagadeesan, R.; Kumar, N.; Saha, S.; Kothandan, G.; Kumar, D. In-silico studies on Myo inositol-1-phosphate synthase of Leishmania donovani in search of anti-leishmaniasis. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Ambrosioni, J.; Blanco, J.L.; Reyes-Urueña, J.M.; Davies, M.-A.; Sued, O.; Marcos, M.A.; Martínez, E.; Bertagnolio, S.; Alcamí, J.; Miro, J.M.; et al. Overview of SARS-CoV-2 infection in adults living with HIV. Lancet HIV 2021, 8, e294–e305. [Google Scholar] [CrossRef]
- Gutierrez, M.D.M.; Mur, I.; Mateo, M.G.; Vidal, F.; Domingo, P. Pharmacological considerations for the treatment of COVID-19 in people living with HIV (PLWH). Expert Opin. Pharmacother. 2021, 1127–1141. [Google Scholar] [CrossRef]
- Nygaard, M.; Kragelund, B.B.; Papaleo, E.; Lindorff-Larsen, K. An Efficient Method for Estimating the Hydrodynamic Radius of Disordered Protein Conformations. Biophys. J. 2017, 113, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Kumar, V.; Meena, N.K.; Lynn, A.M. Elucidation of the structural stability and dynamics of heterogeneous intermediate ensembles in unfolding pathway of the N-terminal domain of TDP-43. RSC Adv. 2018, 8, 19835–19845. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Bagchi, B. Dynamical control by water at a molecular level in protein dimer association and dissociation. Proc. Natl. Acad. Sci. USA 2020, 117, 2302. [Google Scholar] [CrossRef]
- Teli, D.M.; Shah, M.B.; Chhabria, M.T. In silico Screening of Natural Compounds as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Srivastava, R.; Prakash, A.; Lynn, A.M. Structure-based virtual screening, molecular dynamics simulation and MM-PBSA toward identifying the inhibitors for two-component regulatory system protein NarL of Mycobacterium Tuberculosis. J. Biomol. Struct. Dyn. 2020, 38, 3396–3410. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, A.; Kaur, G.; Kumar, M. AVCpred: An integrated web server for prediction and design of antiviral compounds. Chem. Biol. Drug Des. 2017, 89, 74–83. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triazole Based Compounds | Binding Affinity Values (kcal/mol) | No. of H-bonds | H-bonds and Interacting Residues | No. of Other Interactions | Other Interaction and Interacting Residues |
|---|---|---|---|---|---|
| Bemcentinib (DB12411) | −10.2 | 2 | Ser46, Thr26 | 1 | Met49 |
| Bisoctrizole (DB11262) | −9.0 | 2 | Cys44, Gln189 | 1 | Leu50 |
| PYIITM (DB07213) | −8.8 | 4 | His41 (3), Thr45 (1) | 2 | Met49, Cys44 |
| NIPFC (DB07020) | −8.8 | 2 | Cys44, Asn142 | 1 | Met49 |
| Compounds/ Ligands | Water Solubility log mol/L | Caco-2 Permeability log 10−6 cm/s | Human Intestinal Absorption (%) | P-glycoprotein Substrate | P-glycoprotein I Inhibitor | P-glycoprotein II Inhibitor | VDss (log L/kg) | Fraction Unbound (Human) |
|---|---|---|---|---|---|---|---|---|
| Bemcentinib | −3.166 | 1.336 | 100 | Yes | Yes | Yes | 0.755 | 0.179 |
| Bisoctrizole | −2.929 | 1.489 | 100 | No | No | Yes | −1.227 | 0.437 |
| PYIITM | −2.889 | 0.877 | 80.603 | Yes | No | Yes | −0.083 | 0.161 |
| NIPFC | −2.871 | 0.355 | 84.718 | Yes | No | Yes | −0.557 | −0.557 |
| Compounds/ Ligands | CYP2D6 Substrate | CYP3A4 Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|---|
| Bemcentinib (DB12411) | No | Yes | No | Yes | No | No | Yes |
| Bisoctrizole (DB11262) | No | Yes | No | No | No | No | No |
| PYIITM (DB07213) | Yes | Yes | Yes | No | No | No | No |
| NIPFC (DB07020) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Compounds/ Ligands | AMES Toxicity | Total Clearance log ml/ min/kg | Renal OCT2 Substrate | Max. Tolerated Dose (Human) | Oral Rat Acute Toxicity (LD50) | Skin Sensitization | Minnow Toxicity |
|---|---|---|---|---|---|---|---|
| Bemcentinib (DB12411) | Yes | 0.920 | No | 0.181 | 2.995 | No | 1.920 |
| Bisoctrizole (DB11262) | No | −1.185 | No | 0.429 | 3.115 | No | −5.882 |
| PYIITM (DB07213) | No | 1.088 | No | 0.529 | 2.517 | No | 1.985 |
| NIPFC (DB07020) | No | 0.305 | No | 0.602 | 2.890 | No | 3.334 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sur, V.P.; Sen, M.K.; Komrskova, K. In Silico Identification and Validation of Organic Triazole Based Ligands as Potential Inhibitory Drug Compounds of SARS-CoV-2 Main Protease. Molecules 2021, 26, 6199. https://doi.org/10.3390/molecules26206199
Sur VP, Sen MK, Komrskova K. In Silico Identification and Validation of Organic Triazole Based Ligands as Potential Inhibitory Drug Compounds of SARS-CoV-2 Main Protease. Molecules. 2021; 26(20):6199. https://doi.org/10.3390/molecules26206199
Chicago/Turabian StyleSur, Vishma Pratap, Madhab Kumar Sen, and Katerina Komrskova. 2021. "In Silico Identification and Validation of Organic Triazole Based Ligands as Potential Inhibitory Drug Compounds of SARS-CoV-2 Main Protease" Molecules 26, no. 20: 6199. https://doi.org/10.3390/molecules26206199