
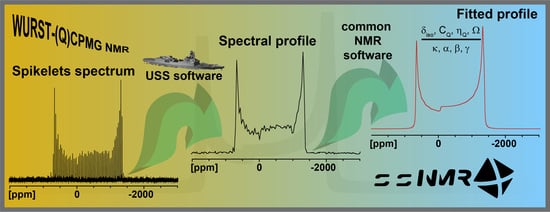
Reconstructing Reliable Powder Patterns from Spikelets (Q)CPMG NMR Spectra: Simplification of UWNMR Crystallography Analysis

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
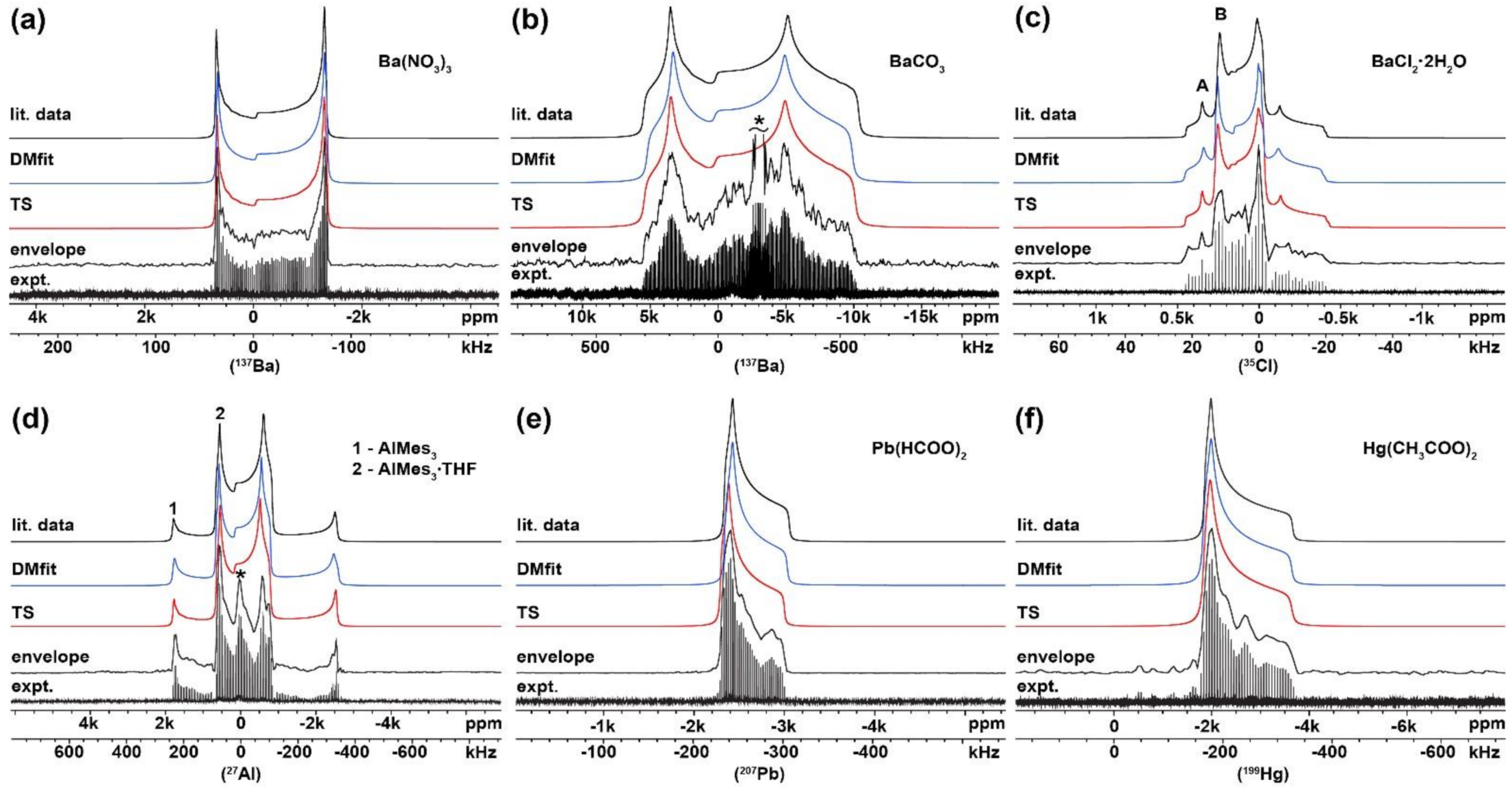
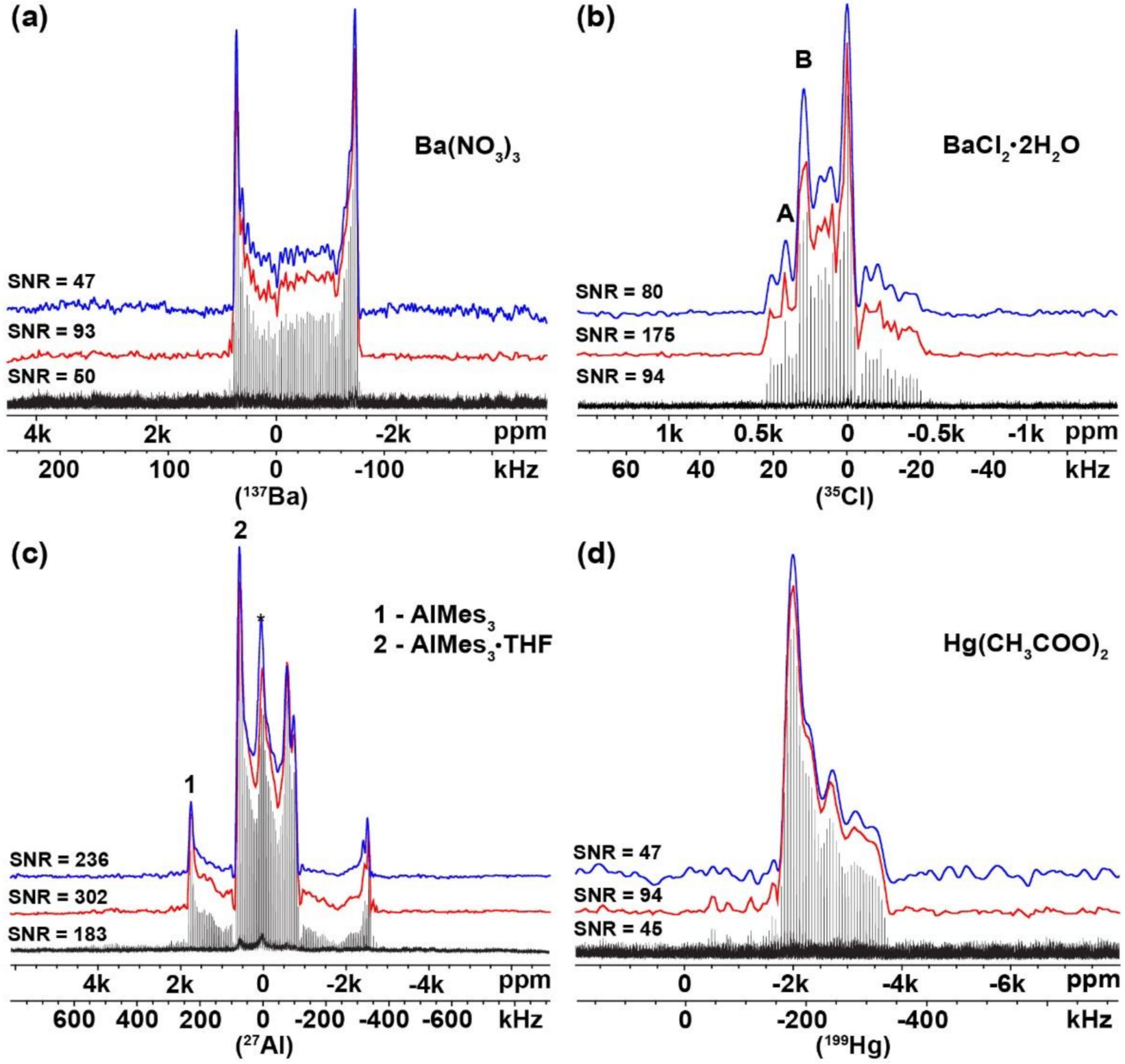
2.1. Verification of the USS Software Using Model Spectra

{kind=link}
{kind=link}
{kind=link}
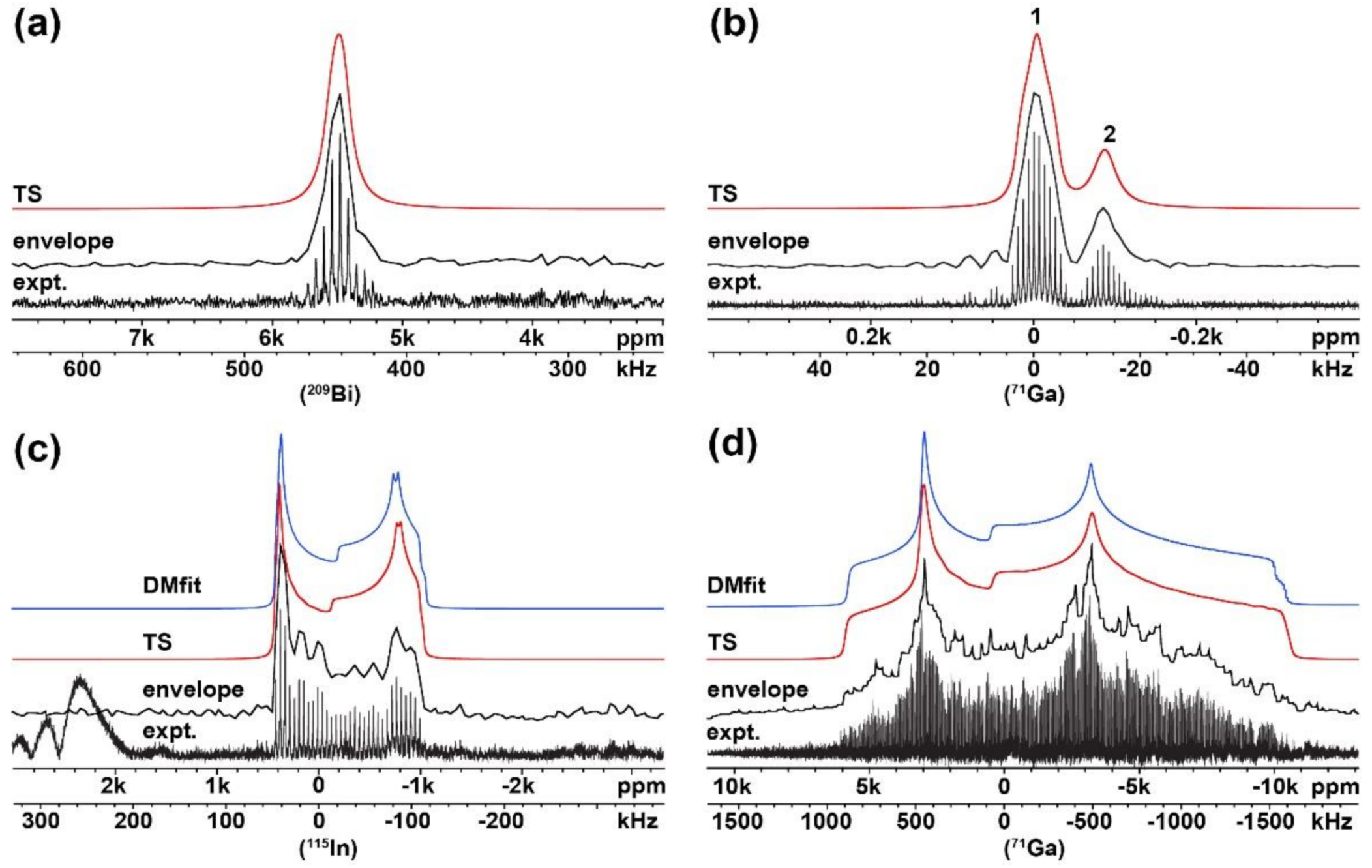{kind=link}
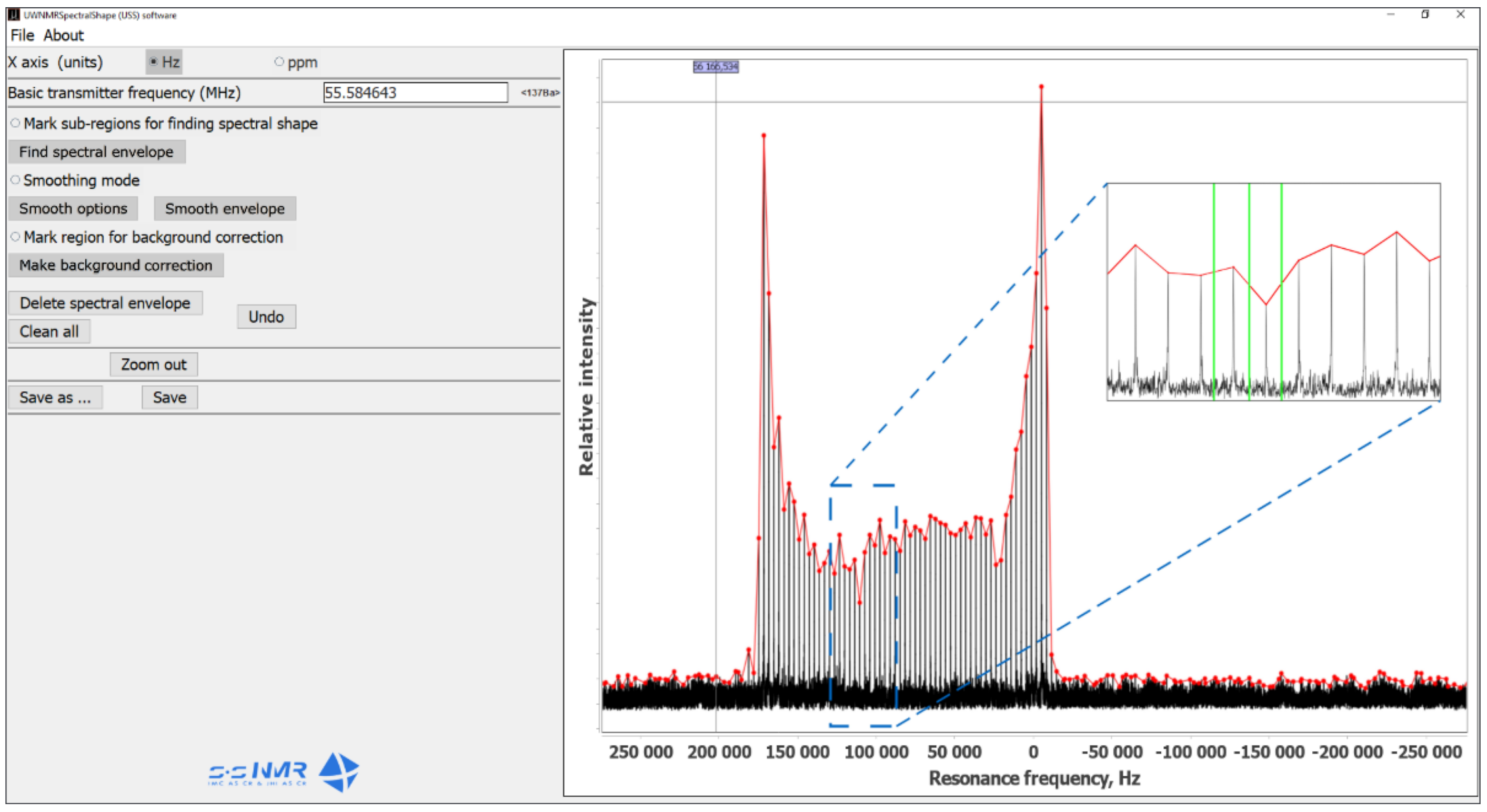{kind=link}
| Compound | Investigated Nuclei | δiso (ppm) | CQ (MHz) | ηQ | Ω (ppm) | κ | α (deg) | β (deg) | γ (deg) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Ba (NO3)2 | 137Ba | −42 (8) | 7.0 (0.10) | 0.01 (0.01) | 25 (20) | 0.80 (0.20) | 40 (20) | 10 (25) | --- | [32] |
| −49 (5) | 6.9 (0.10) | 0.02 (0.01) | 26 (15) | 0.80 (0.20) | 43 (15) | 11 (20) | --- | this work 1 | ||
| −47 (5) | 6.9 (0.10) | 0.02 (0.01) | 24 (15) | 0.70 (0.20) | 53 (20) | 32 (20) | --- | this work 2 | ||
| BaCO3 | 137Ba | 50 (200) | 17.4 (0.60) | 0.33 (0.04) | 150 (150) | 0.50 (0.50) | --- | --- | --- | [32] |
| 76 (150) | 16.8 (0.50) | 0.34 (0.03) | 149 (130) | 0.50 (0.40) | --- | --- | --- | this work 1 | ||
| 108 (150) | 16.6 (0.50) | 0.34 (0.03) | 183 (140) | 0.40 (0.40) | --- | --- | --- | this work 2 | ||
| BaCl2∙2H2O | 35Cl (site A) | 163 (2) | 2.19 (0.08) | 0 | 50 (25) | −0.80 (0.20) | 85 (20) | 32 (10) | 60 (20) | [33] |
| 167 (10) | 2.27 (0.12) | 0 | 39 (25) | −0.60 (0.20) | 89 (20) | 47 (15) | 49 (20) | This work 1 | ||
| 164 (10) | 2.20 (0.12) | 0 | 43 (25) | −0.60 (0.20) | 100 (25) | 30 (15) | 66 (20) | This work 2 | ||
| 35Cl (site B) | 156 (2) | 3.42 (0.08) | 0.30 (0.10) | 50 (25) | 0.20 (0.25) | 20 (15) | 8 (10) | 0 (20) | [33] | |
| 154 (10) | 3.48 (0.15) | 0.30 (0.10) | 52 (25) | 0.20 (0.20) | 22 (15) | 12 (10) | 0 (25) | This work 1 | ||
| 158 (10) | 3.49 (0.15) | 0.30 (0.10) | 47 (25) | 0.10 (0.20) | 15 (15) | 15 (10) | 5 (25) | This work 2 | ||
| AlMes3 | 27Al (1) | 240 (20) | 49.2 (0.50) | 0.01 (0.01) | 126 (10) | −0.99 (0.20) | 180 (20) | 0 (10) | 100 (10) | [30,34] |
| 242 (15) | 49.2 (0.50) | 0.01 (0.01) | 126 (10) | −0.99 (0.20) | 170 (15) | 0 (10) | 110 (10) | this work 1 | ||
| 226 (15) | 48.8 (0.50) | 0.01 (0.01) | 118 (10) | −0.99 (0.20) | 180 (15) | 0 (10) | 100 (10) | this work 2 | ||
| AlMes3∙THF | 27Al (2) | 120 (10) | 27.3 (0.30) | 0.13 (0.10) | 46 (5) | 0.05 (0.10) | 205 (10) | 80 (5) | 200 (10) | [30] |
| 130 (10) | 27.0 (0.30) | 0.17 (0.08) | 47 (5) | 0.05 (0.08) | 195 (10) | 83 (5) | 210 (10) | this work 1 | ||
| 133 (10) | 26.8 (0.30) | 0.17 (0.08) | 51 (5) | 0.07 (0.08) | 200 (10) | 80 (5) | 215 (15) | this work 2 | ||
| Pb (HCOO)2 | 207Pb | −2567 (20) | --- | --- | 720 (20) | 0.75 (0.10) | --- | --- | --- | [35] |
| −2562 (15) | --- | --- | 708 (15) | 0.74 (0.10) | --- | --- | --- | this work 1 | ||
| −2562 (15) | --- | --- | 701 (20) | 0.75 (0.10) | --- | --- | --- | this work 2 | ||
| Hg (CH3COO)2 | 199Hg | -2513 (34) | --- | --- | 1810 (68) | 0.89 (0.07) | --- | --- | --- | [29] |
| −2485 (40) | --- | --- | 1814 (65) | 0.82 (0.07) | --- | --- | --- | this work 1 | ||
| −2488 (40) | --- | --- | 1811 (60) | 0.82 (0.07) | --- | --- | --- | this work 2 |
2.2. Investigation of Compounds with Unknown NMR Parameters
3. Materials and Methods
3.1. Chemicals
3.2. Solid-State NMR
3.3. X-ray Powder Diffraction (XRPD)
3.4. The Periodic Density Functional Theory (DFT) Calculations
3.5. Program Description and Data Processing
- The first point of the region is set as maximum (max)
- FOR all points IN defined region DO (IF intensity of current point is higher than max THEN set current point as max)
- FOR all points IN whole profile (or defined region) DO (IF intensity of the current point is lower than intensities of both (closest) neighboring points THEN intensity of the current point is equal to an average value of intensities of the neighboring points)
- An average intensity (avg) of the first and last points in the defined region is stored.
- FOR all points IN the defined region DO intensity is equal to avg ± 10%.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pake, G.E. Nuclear Resonance Absorption in Hydrated Crystals: Fine Structure of the Proton Line. J. Chem. Phys. 1948, 16, 327–336. [Google Scholar] [CrossRef]
- Pound, R.V. Nuclear Electric Quadrupole Interactions in Crystals. Phys. Rev. 1950, 79, 685–702. [Google Scholar] [CrossRef]
- Smith, J.A.S. Nuclear quadrupole resonance spectroscopy. General principles. J. Chem. Educ. 1971, 48, 39–48. [Google Scholar] [CrossRef]
- Mehring, M. High Resolution NMR Spectroscopy in Solids; Springer: Berlin, Germany, 1976. [Google Scholar]
- Bakhmutov, V.I. Solid-State NMR in Materials Science: Principles and Applications; Paramagnetic Effects in Solid-State NMR; CRC Press: Boca Raton, FL, USA, 2011; p. 191. ISBN 9781439869642. [Google Scholar]
- Samoson, A.; Lippmaa, E.; Pines, A. High resolution solid-state N.M.R. Averaging of second-order effects by means of a double-rotor. Mol. Phys. 1988, 65, 1013–1018. [Google Scholar] [CrossRef]
- Mueller, K.T.; Sun, B.Q.; Chingas, G.C.; Zwanziger, J.W.; Terao, T.; Pines, A. Dynamic-angle spinning of quadrupolar nuclei. J. Magn. Reson. 1990, 86, 470–487. [Google Scholar] [CrossRef]
- Gan, Z. Satellite transition magic-angle spinning nuclear magnetic resonance spectroscopy of half-integer quadrupolar nuclei. J. Chem. Phys. 2001, 114, 10845–10853. [Google Scholar] [CrossRef]
- Huguenard, C.; Taulelle, F.; Knott, B.; Gan, Z. Optimizing STMAS. J. Magn. Reson. 2002, 156, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Medek, A.; Harwood, J.S.; Frydman, L. Multiple-Quantum Magic-Angle Spinning NMR: A New Method for the Study of Quadrupolar Nuclei in Solids. J. Am. Chem. Soc. 1995, 117, 12779–12787. [Google Scholar] [CrossRef]
- Schurko, R.W. Ultra-Wideline Solid-State NMR Spectroscopy. Acc. Chem. Res. 2013, 46, 1985–1995. [Google Scholar] [CrossRef]
- Kobera, L.; Southern, S.A.; Rao, G.K.; Richeson, D.S.; Bryce, D.L. New Experimental Insight into the Nature of Metal−Metal Bonds in Digallium Compounds: J Coupling between Quadrupolar Nuclei. Chem.—A Eur. J. 2016, 22, 9565–9573. [Google Scholar] [CrossRef]
- Kobera, L.; Southern, S.A.; Frost, J.M.; Bryce, D.L. Multinuclear solid-state magnetic resonance study of oxo-bridged diniobium and quadruply-bonded dimolybdenum carboxylate clusters. Solid State Nucl. Magn. Reson. 2017, 84, 20–27. [Google Scholar] [CrossRef]
- Rhodes, H.E.; Wang, P.-K.; Stokes, H.T.; Slichter, C.P.; Sinfelt, J.H. NMR of platinum catalysts. I. Line shapes. Phys. Rev. B 1982, 26, 3559–3568. [Google Scholar] [CrossRef] [Green Version]
- Bastow, T.J.; Smith, M.E. 91Zr NMR characterisation of phases in transformation toughened zirconia. Solid State Nucl. Magn. Reson. 1992, 1, 165–174. [Google Scholar] [CrossRef]
- Massiot, D.; Farnan, I.; Gautier, N.; Trumeau, D.; Trokiner, A.; Coutures, J.P. 71Ga and 69Ga nuclear magnetic resonance study of β-Ga2O3: Resolution of four- and six-fold coordinated Ga sites in static conditions. Solid State Nucl. Magn. Reson. 1995, 4, 241–248. [Google Scholar] [CrossRef]
- Hahn, E.L. Spin Echoes. Phys. Rev. 1950, 80, 580–594. [Google Scholar] [CrossRef]
- Das, T.P.; Saha, A.K. Electric quadrupole interaction and spin echoes in crystals. Phys. Rev. 1955, 98, 516–524. [Google Scholar] [CrossRef]
- Solomon, I. Multiple Echoes in Solids. Phys. Rev. 1958, 110, 61–65. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Shore, S.E.; Ansermet, J.P.; Slichter, C.P.; Sinfelt, J.H. Nuclear magnetic resonance study of the bonding and diffusion of CO chemisorbed on Pd. Phys. Rev. Lett. 1987, 58, 953–956. [Google Scholar] [CrossRef]
- Larsen, F.H.; Jakobsen, H.J.; Ellis, P.D.; Nielsen, N.C. Sensitivity-enhanced quadrupolar-echo NMR of half-integer quadrupolar nuclei. Magnitudes and relative orientation of chemical shielding and quadrupolar coupling tensors. J. Phys. Chem. A 1997, 101, 8597–8606. [Google Scholar] [CrossRef]
- Lipton, A.S.; Wright, T.A.; Bowman, M.K.; Reger, D.L.; Ellis, P.D. Solid-state 67Zn NMR spectroscopy in bioinorganic chemistry. Spectra of four- and six-coordinate zinc pyrazolylborate complexes obtained by management of proton relaxation rates with a paramagnetic dopant. J. Am. Chem. Soc. 2002, 124, 5850–5860. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Frydman, L. Quadrupolar nuclear magnetic resonance spectroscopy in solids using frequency-swept echoing pulses. J. Chem. Phys. 2007, 127, 194503. [Google Scholar] [CrossRef]
- Kupce, E.; Freeman, R. Adiabatic pulses for wideband inversion and broadband decoupling. J. Magn. Reson. Ser. A 1995, 115, 273–276. [Google Scholar] [CrossRef]
- O’Dell, L.A.; Schurko, R.W. QCPMG using adiabatic pulses for faster acquisition of ultra-wideline NMR spectra. Chem. Phys. Lett. 2008, 464, 97–102. [Google Scholar] [CrossRef]
- Tang, J.A.; O’Dell, L.A.; Aguiar, P.M.; Lucier, B.E.G.; Sakellariou, D.; Schurko, R.W. Application of static microcoils and WURST pulses for solid-state ultra-wideline NMR spectroscopy of quadrupolar nuclei. Chem. Phys. Lett. 2008, 466, 227–234. [Google Scholar] [CrossRef]
- MacGregor, A.W.; O’Dell, L.A.; Schurko, R.W. New methods for the acquisition of ultra-wideline solid-state NMR spectra of spin-1/2 nuclides. J. Magn. Reson. 2011, 208, 103–113. [Google Scholar] [CrossRef]
- Kobera, L.; Czernek, J.; Abbrent, S.; Mackova, H.; Pavlovec, L.; Rohlicek, J.; Brus, J. The Nature of Chemical Bonding in Lewis Adducts as Reflected by 27Al NMR Quadrupolar Coupling Constant: Combined Solid-State NMR and Quantum Chemical Approach. Inorg. Chem. 2018, 57, 7428–7437. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.; Gan, Z. On the practical aspects of recording wideline QCPMG NMR spectra. J. Magn. Reson. 2010, 204, 256–265. [Google Scholar] [CrossRef]
- Hamaed, H.; Ye, E.; Udachin, K.; Schurko, R.W. Solid-State 137Ba NMR spectroscopy: An experimental and theoretical investigation of 137Ba electric field gradient tensors and their relation to structure and symmetry. J. Phys. Chem. B 2010, 114, 6014–6022. [Google Scholar] [CrossRef]
- Bryce, D.L.; Bultz, E.B. Alkaline earth chloride hydrates: Chlorine quadrupolar and chemical shift tensors by solid-state NMR spectroscopy and plane wave pseudopotential calculations. Chem.—A Eur. J. 2007, 13, 4786–4796. [Google Scholar] [CrossRef]
- Tang, J.A.; Masuda, J.D.; Boyle, T.J.; Schurko, R.W. Ultra-wideline 27Al NMR investigation of three- and five-coordinate aluminum environments. ChemPhysChem 2006, 7, 117–130. [Google Scholar] [CrossRef]
- Švarcová, S.; Kočí, E.; Bezdička, P.; Garrappa, S.; Kobera, L.; Plocek, J.; Brus, J.; Šťastný, M.; Hradil, D. Uncovering lead formate crystallization in oil-based paintings. Dalt. Trans. 2020, 49, 5044–5054. [Google Scholar] [CrossRef]
- Power, W.P.; Wasylishen, R.E.; Mooibroek, S.; Pettitt, B.A.; Danchura, W. Simulation of NMR powder line shapes of quadrupolar nuclei with half-integer spin at low-symmetry sites. J. Phys. Chem. 1990, 94, 591–598. [Google Scholar] [CrossRef]
- Slavney, A.H.; Hu, T.; Lindenberg, A.M.; Karunadasa, H.I. A Bismuth-Halide Double Perovskite with Long Carrier Recombination Lifetime for Photovoltaic Applications. J. Am. Chem. Soc. 2016, 138, 2138–2141. [Google Scholar] [CrossRef]
- Hamaed, H.; Laschuk, M.W.; Terskikh, V.V.; Schurko, R.W. Application of solid-state 209Bi NMR to the structural characterization of bismuth-containing materials. J. Am. Chem. Soc. 2009, 131, 8271–8279. [Google Scholar] [CrossRef]
- Bieroń, J.; Pyykkö, P. Nuclear quadrupole moments of bismuth. Phys. Rev. Lett. 2001, 87, 133003. [Google Scholar] [CrossRef]
- Leroy, C.; Bryce, D.L. Recent advances in solid-state nuclear magnetic resonance spectroscopy of exotic nuclei. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 109, 160–199. [Google Scholar] [CrossRef]
- Ji, F.; Huang, Y.; Wang, F.; Kobera, L.; Xie, F.; Klarbring, J.; Abbrent, S.; Brus, J.; Yin, C.; Simak, S.I.; et al. Near-Infrared Light-Responsive Cu-Doped Cs2AgBiBr6. Adv. Funct. Mater. 2020, 30, 2005521. [Google Scholar] [CrossRef]
- Knapp, C.E.; Pugh, D.; McMillan, P.F.; Parkin, I.P.; Carmalt, C.J. Synthetic and structural studies of donor-functionalized alkoxy derivatives of gallium. Inorg. Chem. 2011, 50, 9491–9498. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Kobera, L.; Havlin, J.; Abbrent, S.; Rohlicek, J.; Streckova, M.; Sopcak, T.; Kyselova, V.; Czernek, J.; Brus, J. Gallium Species Incorporated into MOF Structure: Insight into the Formation of a 3D Polycrystalline Gallium-Imidazole Framework. Inorg. Chem. 2020, 59, 13933–13941. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B—Condens. Matter Mater. Phys. 2001, 63, 2451011–2451013. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 24401. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Payne, M.C.; Mauri, F. Relativistic nuclear magnetic resonance chemical shifts of heavy nuclei with pseudopotentials and the zeroth-order regular approximation. J. Chem. Phys. 2003, 118, 5746–5753. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA Materials Studio. Dassault Systèmes, Vélizy-Villacoublay: Paris, France. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 29 September 2021).




| Compound | Investigated Nuclei | δiso (ppm) | σiso (ppm) | CQ (MHz) | ηQ | Ω (ppm) | κ | α (deg) | β (deg) | γ (deg) | LB/GB (kHz) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cs2AgBiBr6 * | 207Bi | 4356 | --- * | --- * | --- * | --- * | --- * | --- * | --- * | --- * | DFT | |
| 5517 (25) | --- * | --- * | --- * | --- * | --- * | --- * | --- * | 10.5/9.3 | Expt. 1 | |||
| Ga (NO3)3∙9H2O | 71Ga (1) | 1680 | 1.5 | 0.76 | 68 | 0.76 | 144 | 32 | 310 | --- | DFT | |
| −1 (5) | 1.0 (0.5) | 0.7 (0.10) | 51 (10) | 0.7 (0.2) | 128 (40) | 50 (20) | 293 (20) | 1.5/1.2 | Expt. 1 | |||
| Ga13(NO3)15(OH)24 ∙24H2O | 71Ga (2) | −80 (10) | 0.9 (0.4) | 0.2 (0.1) | 33 (10) | 0.4 (0.2) | 14 (20) | 138 (30) | 177 (25) | 3/0.5 | Expt. 1 | |
| In (NO3)3∙5H2O | 115In | 3745 | 50.4 | 0.19 | 314 | 0.61 | 90 | 20 | 270 | --- | DFT | |
| −80 (10) | 45.7 (0.5) | 0.14 (0.03) | 117 (25) | 0.3 (0.1) | 60 (15) | 10 (10) | 300 (30) | 1.4/0 | Expt. 1 | |||
| −90 (10) | 45.1 (0.5) | 0.15 (0.03) | 130 (25) | 0.4 (0.1) | 62 (15) | 14 (10) | 307 (30) | 1.7/0 | Expt. 2 | |||
| [Cl (Me) Ga (OtBu)]2 | 71Ga | 1408 | 37.8 | 0.53 | 491 | 0.59 | 174 | 10 | 274 | --- | DFT | |
| 286 (23) | 44.3 (0.4) | 0.49 (0.05) | 546 (55) | 0.7 (0.2) | 135 (40) | 12 (10) | 273 (20) | 16.2/17.1 | Expt. 1 | |||
| 307 (24) | 43.7 (0.4) | 0.49 (0.05) | 543 (50) | 0.6 (0.2) | 130 (35) | 18 (10) | 288 (25) | 15.6/0 | Expt. 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahun, A.; Abbrent, S.; Czernek, J.; Rohlicek, J.; Macková, H.; Ning, W.; Konefał, R.; Brus, J.; Kobera, L. Reconstructing Reliable Powder Patterns from Spikelets (Q)CPMG NMR Spectra: Simplification of UWNMR Crystallography Analysis. Molecules 2021, 26, 6051. https://doi.org/10.3390/molecules26196051
Mahun A, Abbrent S, Czernek J, Rohlicek J, Macková H, Ning W, Konefał R, Brus J, Kobera L. Reconstructing Reliable Powder Patterns from Spikelets (Q)CPMG NMR Spectra: Simplification of UWNMR Crystallography Analysis. Molecules. 2021; 26(19):6051. https://doi.org/10.3390/molecules26196051
Chicago/Turabian StyleMahun, Andrii, Sabina Abbrent, Jiri Czernek, Jan Rohlicek, Hana Macková, Weihua Ning, Rafał Konefał, Jiří Brus, and Libor Kobera. 2021. "Reconstructing Reliable Powder Patterns from Spikelets (Q)CPMG NMR Spectra: Simplification of UWNMR Crystallography Analysis" Molecules 26, no. 19: 6051. https://doi.org/10.3390/molecules26196051