Arylaminopropanone Derivatives as Potential Cholinesterase Inhibitors: Synthesis, Docking Study and Biological Evaluation

 and
and
Abstract
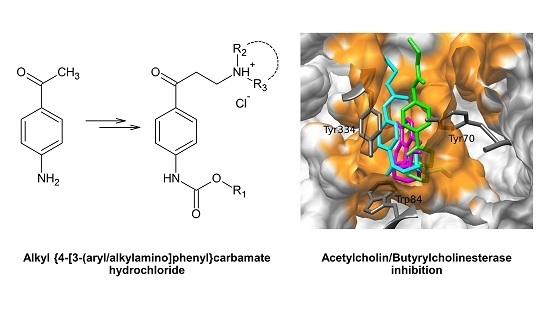
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Enzyme Assays
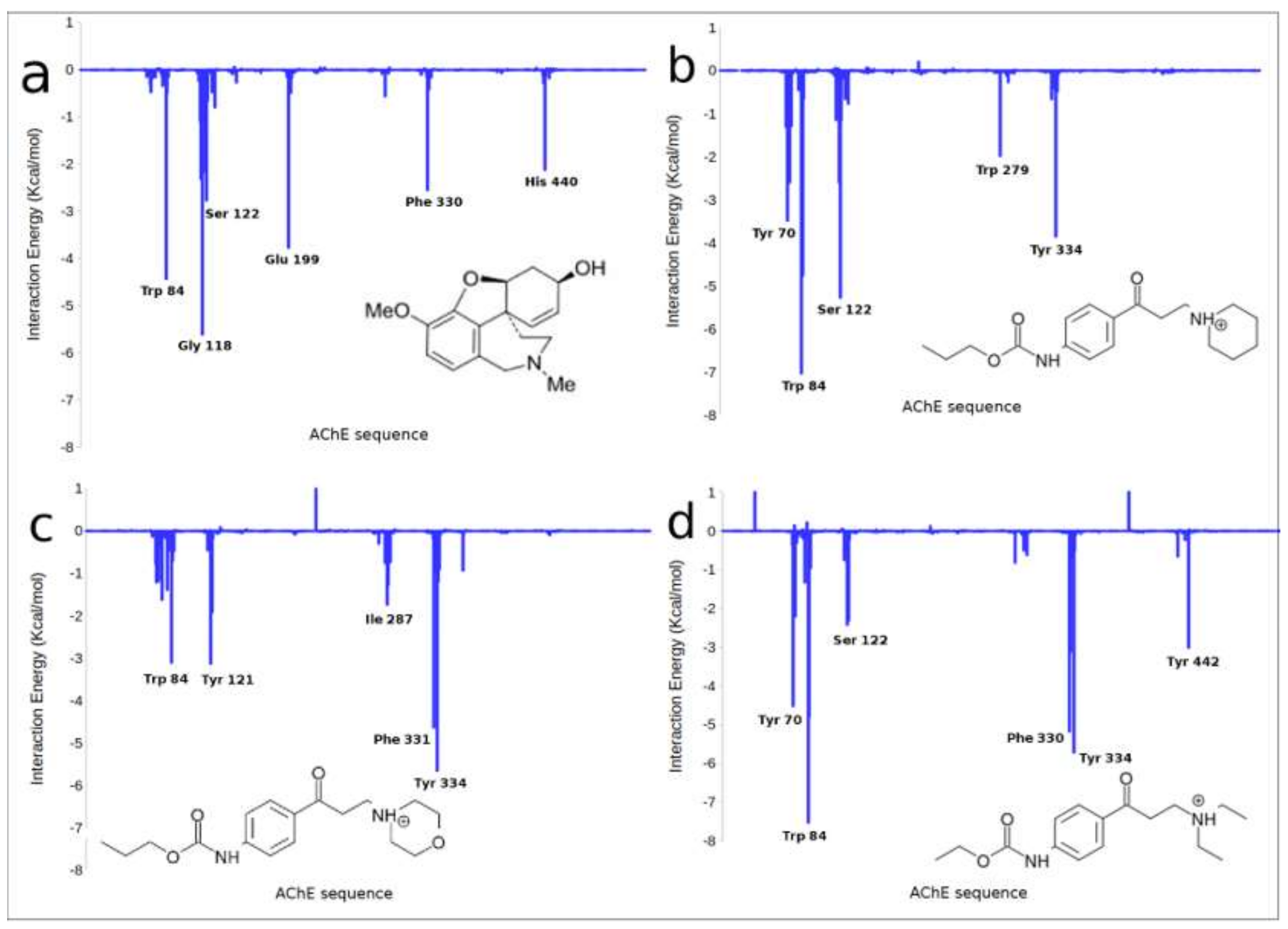
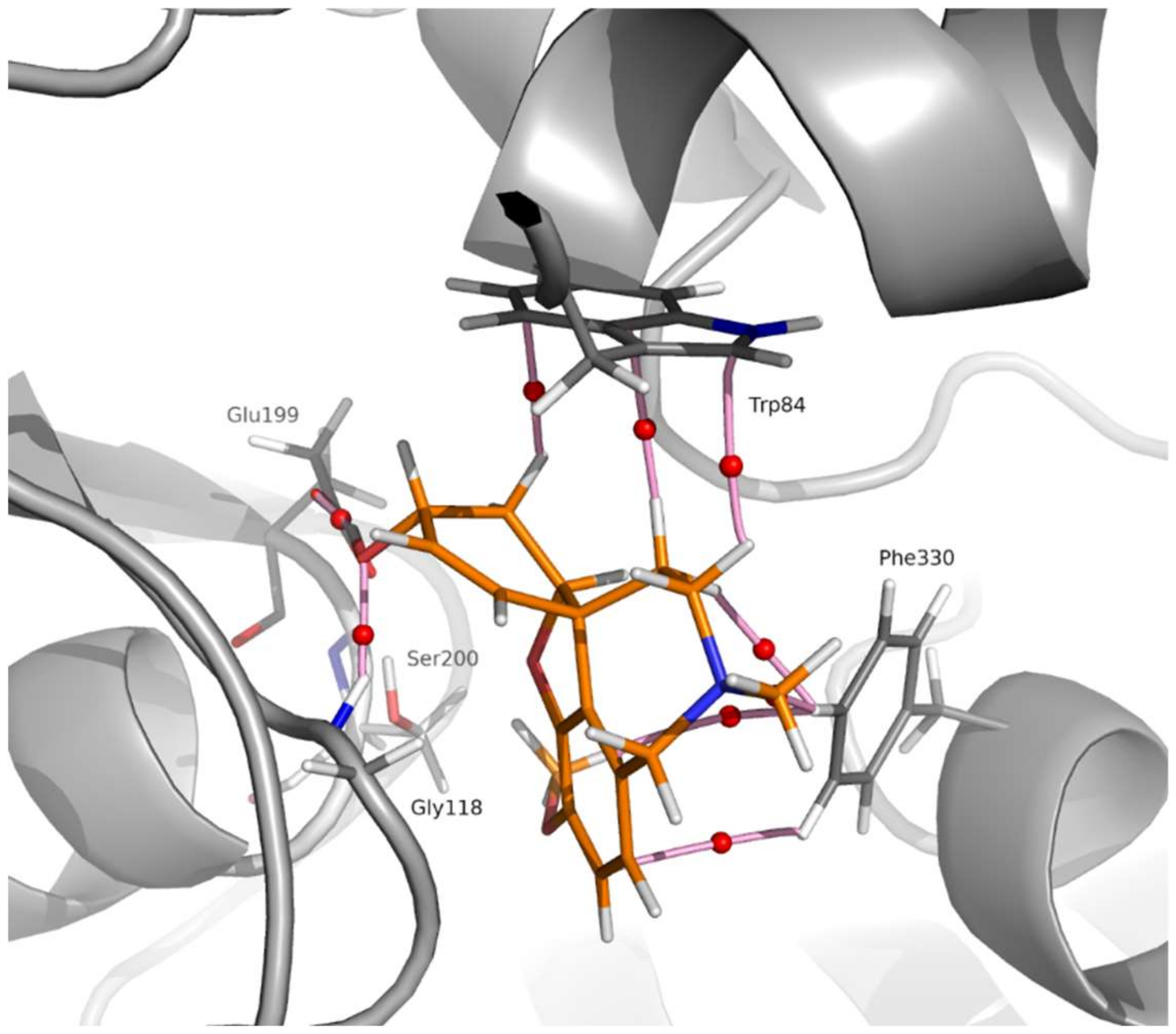
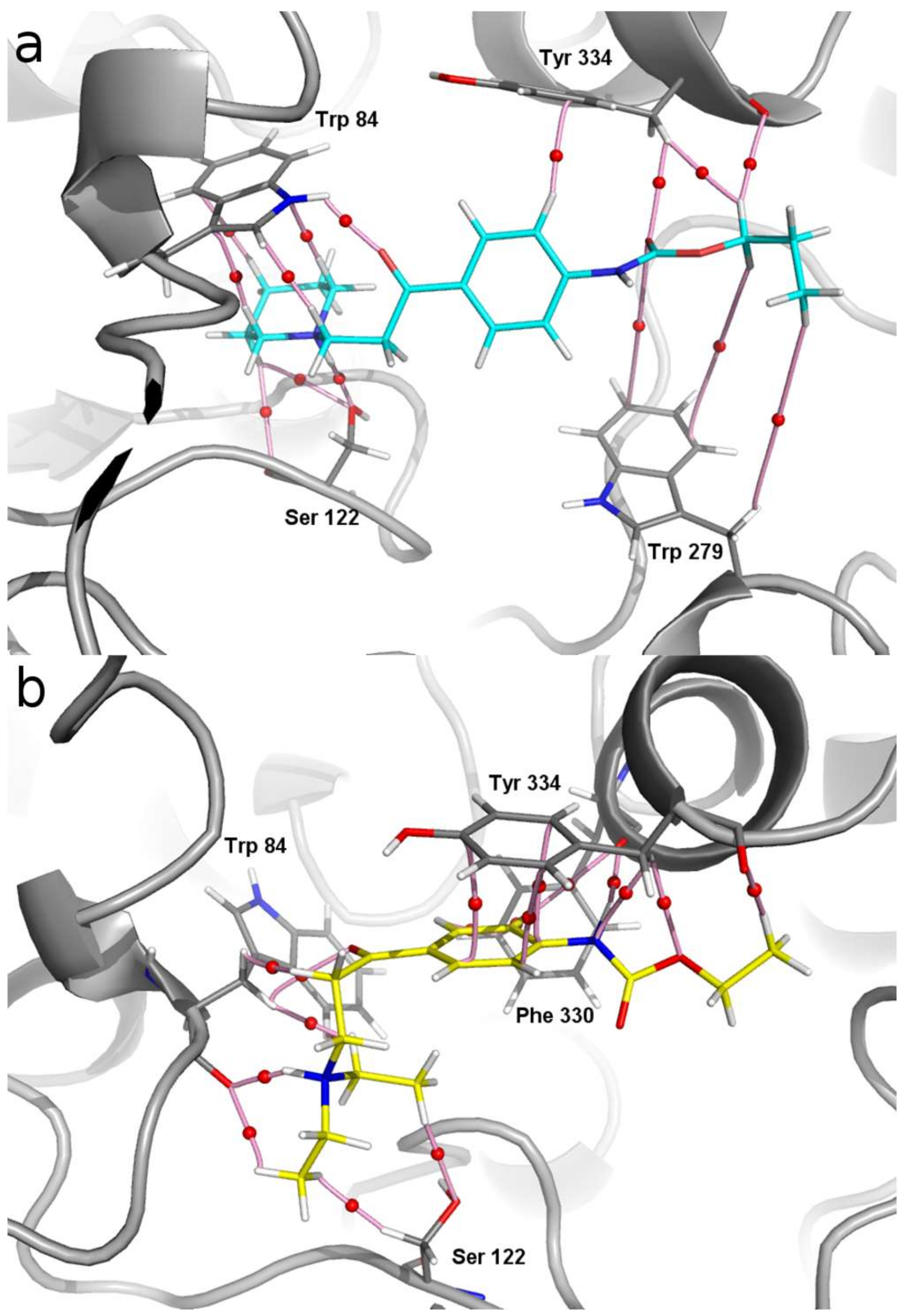
2.3. Molecular Modelling
2.3.1. Docking and MD Simulations
2.3.2. QTAIM Analysis
3. Conclusions
4. Experimental Section
4.1. General Information
4.2. Synthesis
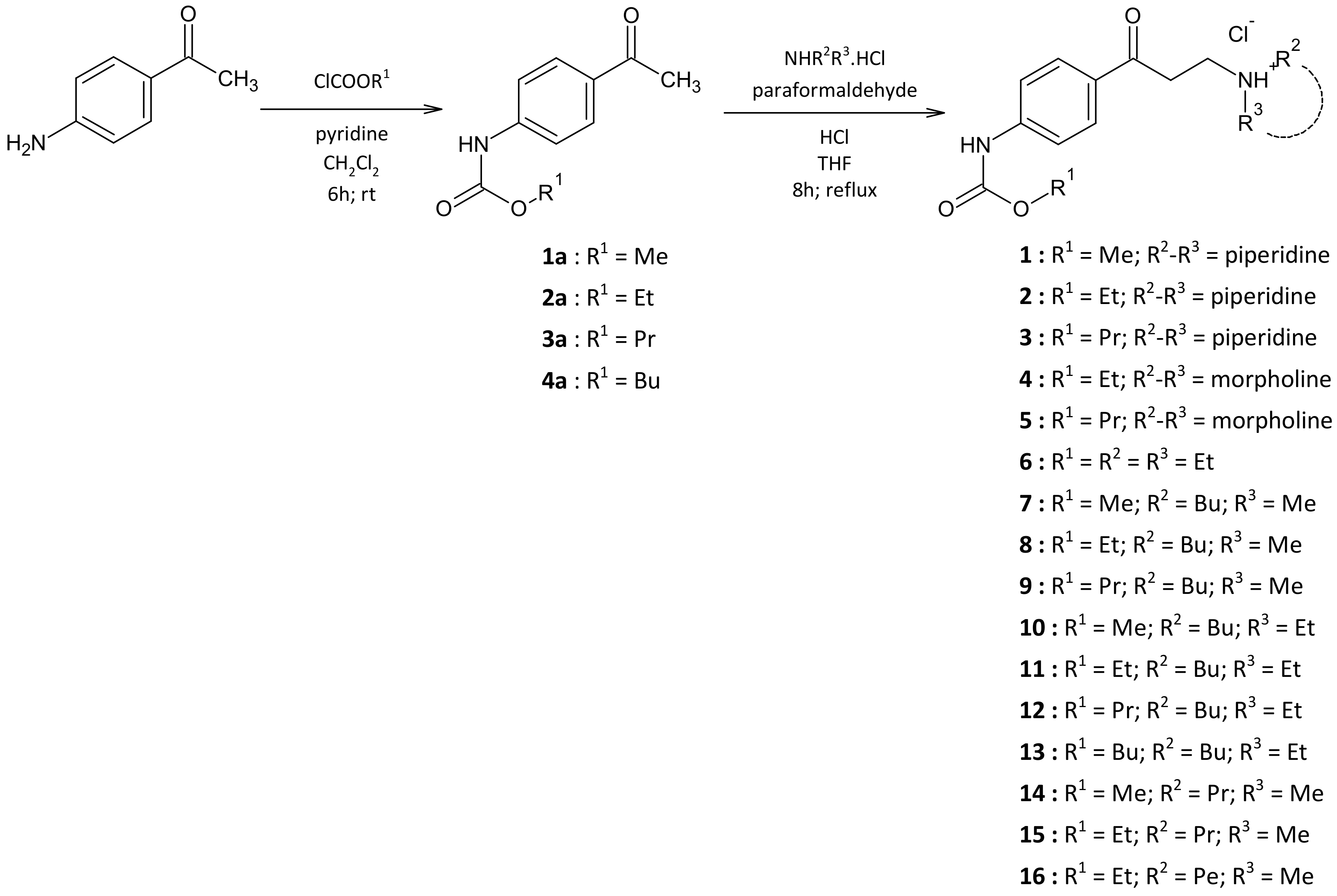
4.2.1. Preparation of Carbamate Intermediates 1a–4a
4.2.2. Preparation of Target Arylaminopropanones 1–16
4.3. Enzyme Assays
4.4. Molecular Modelling
4.4.1. Molecular Modelling
4.4.2. Molecular Docking
4.4.3. Molecular Dynamics (MD) Simulations
4.4.4. Binding Energy Calculations
4.4.5. Atoms in Molecules Theory
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Dementia: A Public Health Priority; World Health Organization: Geneva, Switzerland, 2012; ISBN 9789241564458. [Google Scholar]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A. Acetylcholine. Br. J. Pharmacol. 2006, 147, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, R.L. Biogenesis, assembly and trafficking of acetylcholinesterase. J. Neurochem. 2017, 142, 52–58. [Google Scholar] [CrossRef]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Kryger, G.; Harel, M.; Giles, K.; Toker, L.; Velan, B.; Lazar, A.; Kronman, C.; Barak, D.; Ariel, N.; Shafferman, A.; et al. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Harel, M.; Kryger, G.; Rosenberry, T.L.; Mallender, W.D.; Lewis, T.; Fletcher, R.J.; Guss, J.M.; Silman, I.; Sussman, J.L. Three-dimensional structures of Drosophila melanogaster acetylcholinesterase and of its complexes with two potent inhibitors. Protein Sci. 2000, 9, 1063–1072. [Google Scholar] [CrossRef]
- Šinko, G. Assessment of scoring functions and in silico parameters for AChE-ligand interactions as a tool for predicting inhibition potency. Chem. Biol. Interact. 2019, 308, 216–223. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [Green Version]
- Colletier, J.P.; Fournier, D.; Greenblatt, H.M.; Stojan, J.; Sussman, J.L.; Zaccai, G.; Silman, I.; Weik, M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006, 25, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the Binding of Reversible Inhibitors to Human Butyrylcholinesterase and Acetylcholinesterase: A Crystallographic, Kinetic and Calorimetric Study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sussman, J.L.; Harel, M.; Silman, I. Three-dimensional structure of acetylcholinesterase and of its complexes with anticholinesterase drugs. Chem. Biol. Interact. 1993, 87, 187–197. [Google Scholar] [CrossRef]
- Wu, M.Y.; Esteban, G.; Brogi, S.; Shionoya, M.; Wang, L.; Campiani, G.; Unzeta, M.; Inokuchi, T.; Butini, S.; Marco-Contelles, J. Donepezil-like multifunctional agents: Design, synthesis, molecular modeling and biological evaluation. Eur. J. Med. Chem. 2016, 121, 864–879. [Google Scholar] [CrossRef]
- Harvey, A.L. The pharmacology of galanthamine and its analogues. Pharmacol. Ther. 1995, 68, 113–128. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Bajda, M.; Łątka, K.; Hebda, M.; Jończyk, J.; Malawska, B. Novel carbamate derivatives as selective butyrylcholinesterase inhibitors. Bioorgan. Chem. 2018, 78, 29–38. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [Green Version]
- Vorčáková, K.; Májeková, M.; Horáková, E.; Drabina, P.; Sedlák, M.; Štěpánková, Š. Synthesis and characterization of new inhibitors of cholinesterases based on N-phenylcarbamates: In vitro study of inhibitory effect, type of inhibition, lipophilicity and molecular docking. Bioorgan. Chem. 2018, 78, 280–289. [Google Scholar] [CrossRef]
- Bosak, A.; Smilović, I.G.; Štimac, A.; Vinković, V.; Šinko, G.; Kovarik, Z. Peripheral site and acyl pocket define selective inhibition of mouse butyrylcholinesterase by two biscarbamates. Arch. Biochem. Biophys. 2013, 529, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Darvesh, K.V.; McDonald, R.S.; Mataija, D.; Walsh, R.; Mothana, S.; Lockridge, O.; Martin, E. Carbamates with Differential Mechanism of Inhibition Toward Acetylcholinesterase and Butyrylcholinesterase. J. Med. Chem. 2008, 51, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Bosak, A.; Gazić Smilović, I.; Šinko, G.; Vinković, V.; Kovarik, Z. Metaproterenol, Isoproterenol, and Their Bisdimethylcarbamate Derivatives as Human Cholinesterase Inhibitors. J. Med. Chem. 2012, 55, 6716–6723. [Google Scholar] [CrossRef] [PubMed]
- Kettmann, V.; Csöllei, J.; Račanská, E.; Švec, P. Synthesis and structure-activity relationships of new beta-adrenoreceptor antagonists. Evidence for the electrostatic requirements for beta-adrenoreceptor antagonistrs. Eur. J. Med. Chem. 1991, 26, 843–851. [Google Scholar] [CrossRef]
- Goněc, T.; Malík, I.; Csöllei, J.; Jampílek, J.; Stolaříková, J.; Solovič, I.; Mikuš, P.; Keltošová, S.; Kollár, P.; O’Mahony, J.; et al. Synthesis and In Vitro Antimycobacterial Activity of Novel N-Arylpiperazines Containing an Ethane-1,2-diyl Connecting Chain. Molecules 2017, 22, 2100. [Google Scholar] [CrossRef] [Green Version]
- Fan, P.; Terrier, L.; Hay, A.E.; Marston, A.; Hosttetmann, K. Antioxidant and enzyme inhibition activities and chemical profiles of Polygonum sachalinensis F. Schmidt ex Maxim (Polygonaceae). Fitoterapia 2010, 81, 124–131. [Google Scholar] [CrossRef]
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12 OR; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Bader, R. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Greenblatt, H.M.; Kryger, G.; Lewis, T.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with (−)-galanthamine at 2.3 A resolution. FEBS Lett. 1999, 463, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, J.; Garro, A.; Pigni, N.; Agüero, M.B.; Roitman, G.; Slanis, A.; Enriz, R.D.; Feresin, G.E.; Bastida, J.; Tapia, A. Colinesterase-inhibitory effect and in silico analysis of alkaloids from bulbs of Hieronymiella species. Phytomedicine 2018, 39, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.; Dror, R.; Shaw, D. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.; Caldwell, J.; Kollman, P.; Case, D. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, J.; Catarello, D.; Wozniak, J.; Skeel, R. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015.
- Padrtova, T.; Marvanova, P.; Odehnalova, K.; Kubinova, R.; Parravicini, O.; Garro, A.; Enriz, R.D.; Humpa, O.; Oravec, M.; Mokry, P. Synthesis, Analysis, Cholinesterase-Inhibiting Activity and Molecular Modelling Studies of 3-(Dialkylamino)-2-hydroxypropyl 4-[(Alkoxy-carbonyl)amino]benzoates and Their Quaternary Ammonium Salts. Molecules 2017, 22, 2048. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Andujar, S.A.; Tosso, R.D.; Suvire, F.; Angelina, E.; Peruchena, N.; Cabedo, N.; Cortes, D.E.; Enriz, R.D. Searching the “Biological Relevant” Conformation of Dopamine: A Computational Approach. J. Chem. Inf. Model. 2012, 52, 99–112. [Google Scholar] [CrossRef]
- Tosso, R.D.; Andujar, S.A.; Gutierrez, L.; Angelina, E.; Rodriguez, R.; Nogueras, M.; Baldoni, H.; Suvire, F.D.; Cobo, J.; Enriz, R.D. Molecular modeling study of dihydrofolate reductase inhibitors. Molecular dynamics simulations, quantum mechanical calculations, and experimental corroboration. J. Chem. Inf. Model. 2013, 53, 2018–2032. [Google Scholar] [CrossRef]
- Parraga, J.; Andujar, S.A.; Rojas, S.; Gutierrez, L.J.; El Aouad, N.; Sanz, M.J.; Enriz, D.; Cabedo, N.; Cortes, D. Dopaminergic isoquinolines with hexahydrocyclopenta[ij]-isoquinolines as D 2 -like selective ligands. Eur. J. Med. Chem. 2016, 122, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Parraga, J.; Cabedo, N.; Andujar, S.A.; Piqueras, L.; Moreno, L.; Galan, A.; Angelina, E.; Enriz, D.; Ivorra, M.D.; Sanz, M.J.; et al. 2,3,9- and 2,3,11-Trisubstituted tetrahydroprotoberberines as D 2 dopaminergic ligands. Eur. J. Med. Chem. 2013, 68, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Angelina, E.; Andujar, S.A.; Tosso, R.D.; Enriz, R.D.; Peruchena, N. Non-covalent interactions in receptor-ligand complexes. A study based on the electron charge density. J. Phys. Org. Chem. 2014, 27, 128–134. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–16 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | AChEa (%) | AChE IC50 (µM) | BuChEa (%) | BuChE IC50 (µM) |
| 1 | Me | piperidine | 74.1 ± 0.9 | 12.7 | 10.0 ± 0.8 | >100 | |
| 2 | Et | piperidine | 77.8 ± 0.4 | 8.58 | 15.7 ± 1.9 | >100 | |
| 3 | Pr | piperidine | 79.4 ± 0.3 | 6.57 | 21.0 ± 2.5 | >100 | |
| 4 | Et | morpholine | 16.1 ± 4.2 | >100 | 17.8 ± 3.4 | >100 | |
| 5 | Pr | morpholine | 25.9 ± 3.2 | >100 | 14.4 ± 2.4 | >100 | |
| 6 | Et | Et | Et | 42.2 ± 4.3 | >100 | 34.2 ± 2.6 | >100 |
| 7 | Me | Bu | Me | 38.0 ± 4.2 | >100 | 20.4 ± 5.8 | >100 |
| 8 | Et | Bu | Me | 23.5 ± 2.2 | >100 | 4.7 ± 3.0 | >100 |
| 9 | Pr | Bu | Me | 44.5 ± 1.1 | >100 | 29.0 ± 2.9 | >100 |
| 10 | Me | Bu | Et | 23.6 ± 2.3 | >100 | 43.6 ± 3.0 | >100 |
| 11 | Et | Bu | Et | 24.6 ± 1.9 | >100 | 51.0 ± 2.9 | >100 |
| 12 | Pr | Bu | Et | 38.4 ± 1.3 | >100 | 51.1 ± 3.3 | >100 |
| 13 | Bu | Bu | Et | 33.5 ± 3.4 | >100 | 48.7 ± 2.6 | >100 |
| 14 | Me | Pr | Me | 42.2 ± 1.3 | >100 | 22.3 ± 4.6 | >100 |
| 15 | Et | Pr | Me | 45.3 ± 3.4 | >100 | 36.8 ± 2.1 | >100 |
| 16 | Et | Pe | Me | 50.6 ± 1.8 | >100 | 46.3 ± 2.0 | >100 |
| Galantamine | 87.9 ± 0.5 | 1.1 | 62.1 ± 1.4 | 73.6 | |||
| Rivastigmine | 54.8 ± 4.4 | 87.9 | 72.9 ± 1.5 | 11.6 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudcová, A.; Kroutil, A.; Kubínová, R.; Garro, A.D.; Gutierrez, L.J.; Enriz, D.; Oravec, M.; Csöllei, J. Arylaminopropanone Derivatives as Potential Cholinesterase Inhibitors: Synthesis, Docking Study and Biological Evaluation. Molecules 2020, 25, 1751. https://doi.org/10.3390/molecules25071751
Hudcová A, Kroutil A, Kubínová R, Garro AD, Gutierrez LJ, Enriz D, Oravec M, Csöllei J. Arylaminopropanone Derivatives as Potential Cholinesterase Inhibitors: Synthesis, Docking Study and Biological Evaluation. Molecules. 2020; 25(7):1751. https://doi.org/10.3390/molecules25071751
Chicago/Turabian StyleHudcová, Anna, Aleš Kroutil, Renata Kubínová, Adriana D. Garro, Lucas J. Gutierrez, Daniel Enriz, Michal Oravec, and Jozef Csöllei. 2020. "Arylaminopropanone Derivatives as Potential Cholinesterase Inhibitors: Synthesis, Docking Study and Biological Evaluation" Molecules 25, no. 7: 1751. https://doi.org/10.3390/molecules25071751