 Petra Hyroššová1
Petra Hyroššová1 Mirko Milošević1,2Josef Škoda1
Mirko Milošević1,2Josef Škoda1 Jiří Vachtenheim Jr3
Jiří Vachtenheim Jr3 Jakub Rohlena1*
Jakub Rohlena1* Kateřina Rohlenová1*
Kateřina Rohlenová1*- 1Institute of Biotechnology of the Czech Academy of Sciences, Prague, Czechia
- 2Faculty of Science, Charles University, Prague, Czechia
- 33rd Department of Surgery, First Faculty of Medicine, Charles University and University Hospital Motol, Prague, Czechia
Targeting tumor metabolism for cancer therapy is an old strategy. In fact, historically the first effective cancer therapeutics were directed at nucleotide metabolism. The spectrum of metabolic drugs considered in cancer increases rapidly – clinical trials are in progress for agents directed at glycolysis, oxidative phosphorylation, glutaminolysis and several others. These pathways are essential for cancer cell proliferation and redox homeostasis, but are also required, to various degrees, in other cell types present in the tumor microenvironment, including immune cells, endothelial cells and fibroblasts. How metabolism-targeted treatments impact these tumor-associated cell types is not fully understood, even though their response may co-determine the overall effectivity of therapy. Indeed, the metabolic dependencies of stromal cells have been overlooked for a long time. Therefore, it is important that metabolic therapy is considered in the context of tumor microenvironment, as understanding the metabolic vulnerabilities of both cancer and stromal cells can guide new treatment concepts and help better understand treatment resistance. In this review we discuss recent findings covering the impact of metabolic interventions on cellular components of the tumor microenvironment and their implications for metabolic cancer therapy.
Introduction
Metabolic anti-cancer drugs target pathways such as glycolysis, glutaminolysis or oxidative phosphorylation that support proliferation and redox balance in cancer cells. These pathways are fundamental to cellular metabolism in general, and while being crucial for cancer cells, they are not strictly cancer specific. Non-transformed proliferating cells and to some degree also non-proliferating somatic cells engage the very same pathways to perform a number of essential functions (1). Metabolic drugs that target these pathways will thus also impact cell types other than cancer cells when applied in vivo. Indeed, besides cancer cells, tumors comprise multiple other non-transformed cell types such as fibroblasts, endothelial cells (ECs) and immune cells that collectively form the tumor microenvironment (TME) (Figure 1). Within the TME, cancer cells interact with and alter metabolism of these tumor resident non-transformed stromal cell types on multiple levels. For example, cancer associated fibroblasts (CAFs) upregulate collagen synthesis for extracellular matrix deposition (2, 3), tumor associated ECs stimulate glycolysis to support angiogenesis, and various types of the immune cells such as macrophages or T-cells are modulated by the TME towards immunosuppression. Vice versa, stromal cells may supply cancer cells with metabolites, and cancer cells can obtain nutrients externally from the blood stream (4), which is facilitated by EC-driven angiogenesis.
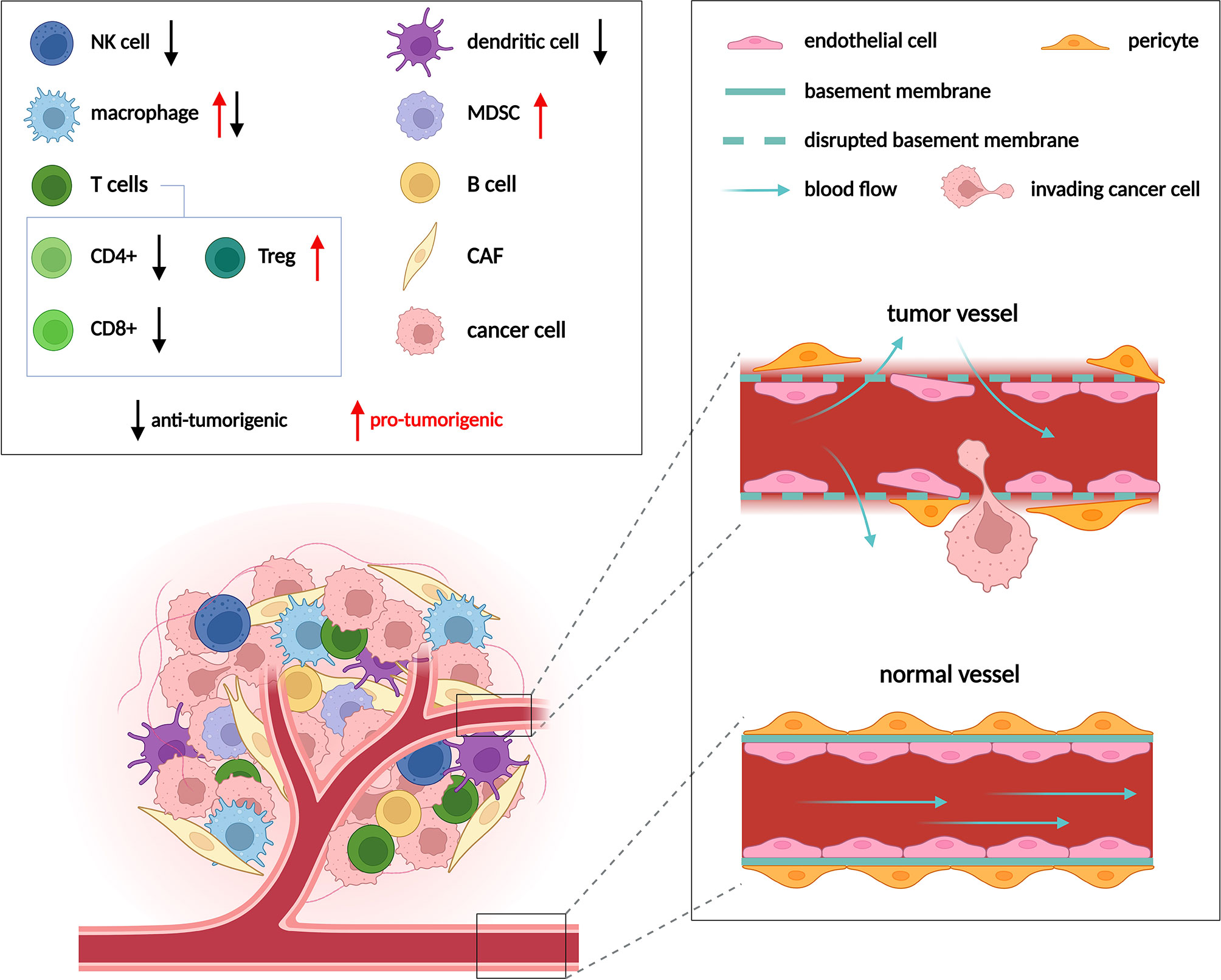
Figure 1 Cell populations in the TME and their role in tumor progression. Composition of the TME varies between different tumor types, but usually includes cancer cells, immune cells, cancer associated fibroblasts (CAFs) and endothelial cells (ECs). Multiple types of immune cells are present in the TME, only those mentioned in following figures are depicted. Immune cells can either promote (red arrows) or suppress (black arrows) tumor growth. ECs form the inner lining of blood vessels. Vessels are normally well ordered, covered by an intact basement membrane and a layer of pericytes. In the TME vessels become disorganized due to excessive pro-angiogenic signaling. Such abnormal vessels are leaky and become a gateway for tumor metastasis. Adapted from “Tumor Microenvironment 2”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
Strategies to target metabolism of tumor cells do not routinely consider the metabolism of non-cancer cells (e.g., immune cells, ECs and CAFs) within the tumor. However, these additional aspects might be essential when searching for a truly effective and practicable metabolic treatment. Below we summarize how pharmacological modulation of major metabolic pathways in cancer affects individual stromal cell types.
Glycolysis
Increased glycolysis is the best known metabolic hallmark of cancer (5, 6). It provides cancer cells with ATP and metabolic intermediates that are channeled into biosynthetic pathways to form nucleotides, amino acids (AAs) and lipids, and to maintain redox homeostasis (7, 8). Pyruvate produced by glycolysis is converted to lactate to sustain high glycolytic flux (9). This conversion is favored because pyruvate import into mitochondria is suppressed by pyruvate dehydrogenase kinase (Figure 2). Attenuation of glycolysis in cancer cells will therefore compromise biosynthesis and alter the redox balance. Two main approaches have been pursued to achieve these goals: (i) direct inhibition/modulation of the glycolytic pathway and (ii) interference with the excretion of lactate.
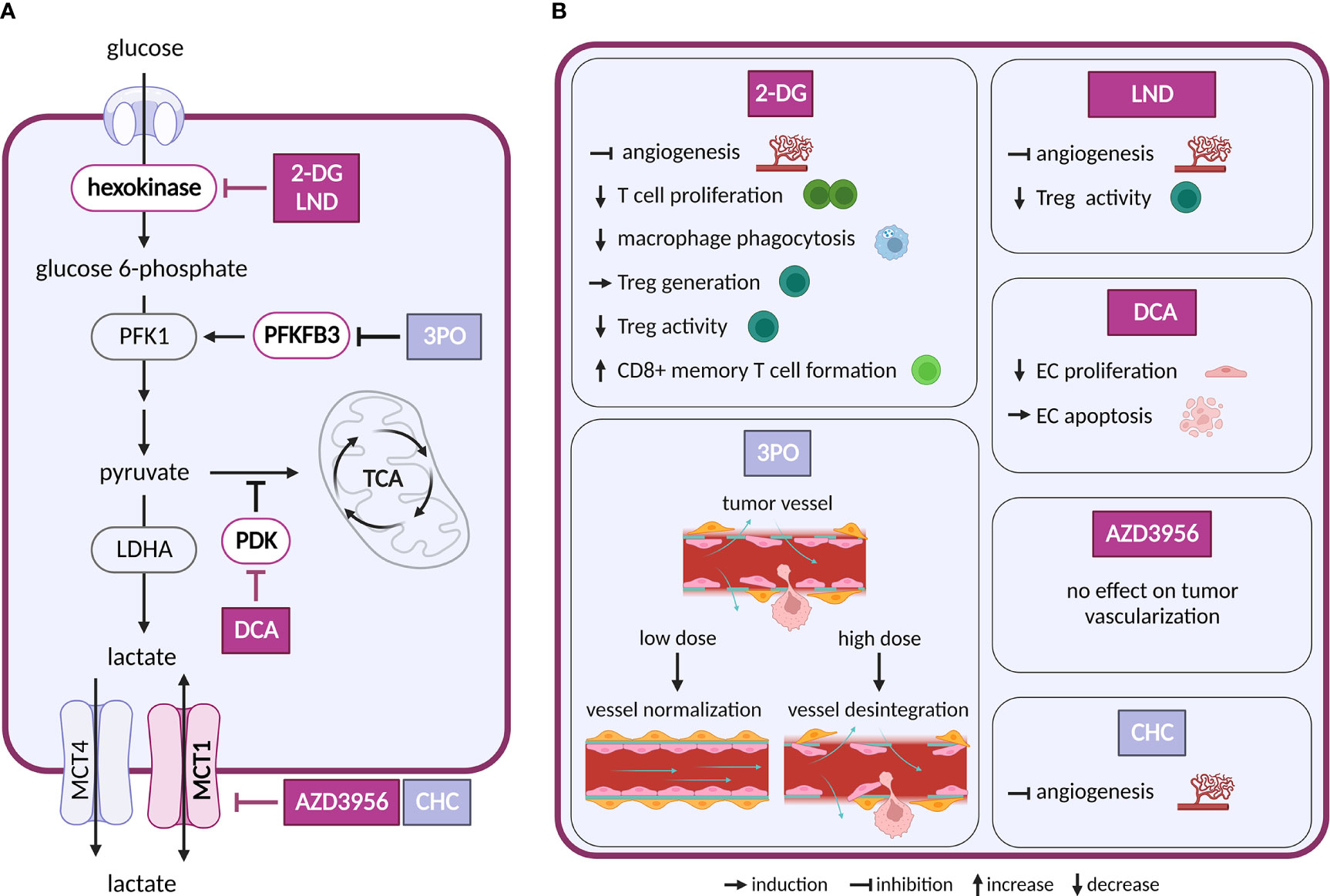
Figure 2 Inhibitors of glycolysis and their effect on TME cell types. (A) Schematic representation of glycolysis and lactate exchange. Only inhibitors with a reported effect in the TME are shown. Inhibitors tested in clinical trials for cancer are highlighted in dark violet, inhibitors tested in preclinical studies are shown in light grey-blue (B) Effects of the inhibitors shown in (A) on cell populations in the TME.
Intervention strategies that inhibit glycolysis are challenging due to its systemic importance. First attempts to target glycolysis by unmetabolizable glucose analog 2-deoxyglucose (2-DG) were unsuccessful, as the treatment caused toxicity associated with hypoglycemia (10). Despite the initial disappointment, other glycolysis inhibitors have been intensively evaluated and several of them were brought into clinical trials (11). The tested drugs target various steps of the glycolytic pathway, such as glucose transporters (GLUTs), hexokinase 2 (HK2), pyruvate dehydrogenase kinase (PDK), lactate dehydrogenase A (LDHA) and monocarboxylate transporters (MCTs).
Besides cancer cells, glycolysis is crucial also in other cell types of the TME. For example, CAFs, an abundant stromal type in solid tumors, display increased expression of enzymes involved in glycolysis and produce lactate (12). Interestingly, redirection of glucose metabolism toward tricarboxylic acid (TCA) cycle in CAFs by silencing PDK4 showed decreased growth of co-injected cancer cells in xenograft models (13).
ECs are glycolytic already at baseline, and their glycolytic flux is further increased in response to tumor-produced vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) (14). Both VEGF and FGF upregulate an activator of glycolysis 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) in tumor ECs (TECs), while FGF also activates a glycolytic enzyme HK2 (15, 16). PFKFB3 is responsible for the production of fructose-2,6-bisphosphate, an allosteric activator of phosphofructokinase 1 (PFK-1). As heightened glycolysis in ECs promotes angiogenesis, inhibition of PFKFB3 or HK2 genetically or pharmacologically, using 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) or 2-DG and lonidamine (LND), respectively, impairs vessel formation in vitro and in vivo (15–19). Interestingly, in the context of tumor angiogenesis inhibition of PFKFB3 by 3PO can have different outcomes, depending on the administered dose. Low dose 3PO, that does not affect cancer cells, leads to normalization of tumor vasculature and consequently to decreased cancer cell invasion and metastasis (20). However, highest tolerable dose of 3PO, used in clinical trials, disrupts TECs metabolism and induces apoptosis, which in turn leads to vessel disintegration and increased metastasis (21). Similar dose-dependence has been observed for dichloroacetate (DCA). This compound, an inhibitor of PDK, promotes shunting of pyruvate into mitochondria, which attenuates aerobic glycolysis. At lower doses DCA decrease proliferation and migration of ECs, but at higher doses it drives ECs to apoptosis (22). It is thus important to routinely evaluate dosing of new glycolytic inhibitors to avoid adverse effects on vessel integrity that might be pro-metastatic.
Glucose is also critical for proliferation and activation of immune cells, such as effector T cells (23), natural killer (NK) cells (24) and M1 macrophages (25). 2-DG decreases phagocytosis of elicited macrophages (26), proliferation of T cells in vitro (27) and reduces IFNγ secretion in splenocytes (28). Depletion of glucose in tumor environment by cancer cells limits effector function of metabolically restricted T cells and leads to their functional exhaustion (29, 30). 2-DG treatment promoted generation of regulatory T cells (Treg) cells in vitro and in vivo in the context of autoimmune disease (31). However, in other study, 2-DG and LND showed opposite results, where it reversed suppressive activity of Treg cells (32). These differences might be caused by different experimental settings or metabolic state of Treg cells and need to be addressed further. Additionally, inhibition of glycolysis with 2-DG promotes formation of long-lived CD8+ T memory cells (33). Overall, considering different metabolic dependencies of immune cell types, it is not surprising that inhibition of glycolysis has different effects on immune response.
The main product of aerobic glycolysis is lactate, an important carbon source not only in the TME, but also during normal physiology (4). Lactate exchange with the extracellular space is maintained by MCT1 and 4 (34). MCT1 allows bi-directional transport that depends on substrate gradient (35). Inhibition of MCT1 is thus expected to interfere with lactate export in highly glycolytic cancer cells, resulting in their elimination. At the same time, this treatment would deprive other cell types of an important carbon source (possibly also inducing cell death). Inhibition or a decrease in expression of MCT1, MCT4 and LDHA in cancer cells slowed down tumor growth (36, 37). Importantly, MCT1 and MCT4 are expressed not only in cancer cells of various origin, but also in stromal cells (35). MCT1 inhibitor AZD3965 underwent Phase I clinical study (NCT01791595).
Several studies suggest a so-called reverse Warburg effect, where CAFs feed cancer cells with lactate or pyruvate that cancer cells oxidize in mitochondria (13, 38, 39). In this setting, silencing of MCT4 in CAFs decreased lactate export from CAFs and decreased growth of tumors in vivo (39). Such reduction in CAF-exported lactate would not only deprive cancer cells of an alternative fuel, but also eliminate the immunosuppressive effects of elevated lactate concentrations (see below).
In ECs, lactate uptake through MCT1 activates hypoxia-inducible factor 1 alpha (HIF-1α) and consequently promotes angiogenesis through increased expression of basic fibroblast growth factor and vascular endothelial growth factor receptor 2 (VEGFR2) (40). However, an inhibition of MCT1 showed different outcomes with two different compounds: a-cyano-4-hydroxycinnamate (CHC) decreased angiogenesis in tumors (40), whereas AZD3965 showed no effect on tumor vascularization in a murine model. These differences might be caused by unspecific effects of CHC (41).
Lactate negatively affects function and survival of T cells and NK cells in the TME, which leads to immune escape. In line with this, lower lactate production in melanoma tumors with downregulated LDHA expression increased infiltration of T and NK cells, which slowed down tumor growth (42). Lactate is also known to promote M2-like polarization of tumor-associated macrophages (TAMs) that promote tumor progression by production of immunosuppressive cytokines (43, 44). Suppression of lactate production in cancer cells lowered the lactate effect on M2 macrophage polarization (45, 46). Of note, in T cells MCT1 inhibition was considered as an immunosuppressive therapy. While MCT1 inhibitors can block T cell proliferation, this does not interfere with their effector function and viability (47).
In summary, while complete inhibition of glycolysis will reduce viability/proliferative potential of tumor cells, it will also suppress anti-tumor immune responses and lead to tumor vessel disintegration that promotes metastasis, rendering treatment ineffective. Partial inhibition of glycolysis might thus result in a more favorable outcome in vivo. On the other hand, inhibition of lactate transporters seems to have less negative effect on the vasculature and immune responses within the TME.
Oxidative phosphorylation
Oxidative phosphorylation (OXPHOS) is a system of five respiratory complexes at the inner membrane of mitochondria that is best known for ATP production (Figure 3). However, in proliferating cancer cells ATP generation by OXPHOS is dispensable, and OXPHOS maintains intracellular redox balance to enable synthesis of AAs aspartate and asparagine as well as de novo synthesis of nucleotides (48–51). OXPHOS activity is often but not always reduced in cancer cell lines and tumors, as an outcome of lower mitochondrial DNA (mtDNA) content or presence of mtDNA mutations (52). Still, even low levels of OXPHOS activity can support biosynthesis and promote primary tumors (53). Interestingly, elevated OXPHOS activity marks drug-resistant and persistent populations of cancer cells (54), and some cancer types upregulate OXPHOS (52, 55), such as those localized in well perfused and oxygenated areas (56, 57). Thus, OXPHOS inhibitors are considered for cancer therapy as they interfere with biosynthesis and upregulate reactive oxygen species (ROS) production from the respiratory chain (58, 59).
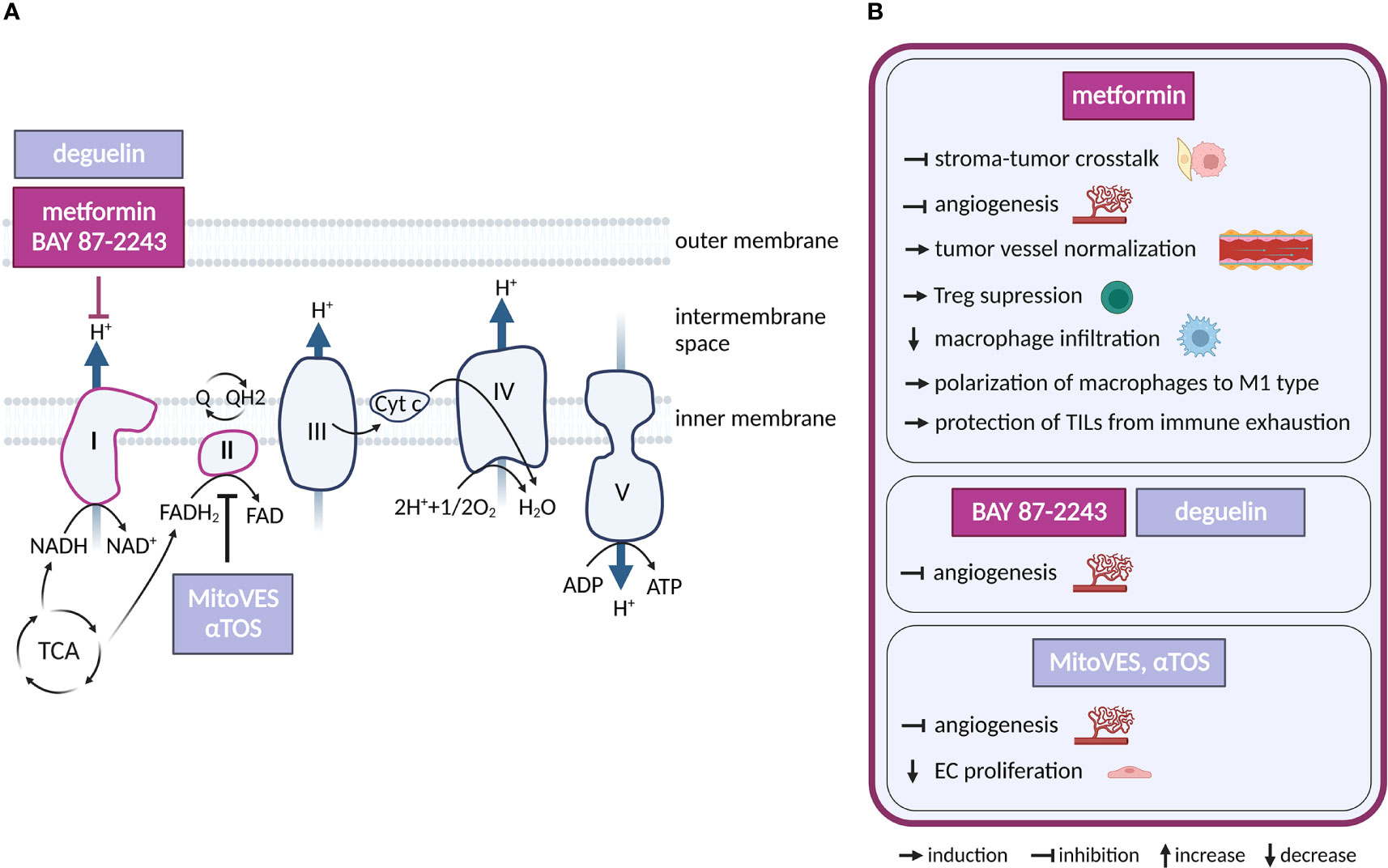
Figure 3 Inhibitors of oxidative phosphorylation and their effects on TME cell types. (A) Schematic representation of the OXPHOS system. Only inhibitors with a reported effect in the TME are shown. Inhibitors tested in clinical trials for cancer and in clinical practice are highlighted in dark violet, those in preclinical studies are in light grey-blue. (B) Effects of the OXPHOS inhibitors shown in (A) on cell populations present in the TME.
Regarding potential cancer therapeutics, respiratory complex I (CI) is the most frequent target. Multiple CI inhibitors with anti-cancer properties have been developed and/or tested, including IACS-010759, BAY 87-2243, tamoxifen, deguelin and the anti-diabetics metformin and phenformin (60–65). Complex II (LND, αTOS) (66, 67), Complex III (atovaquone, adaphostin) (68, 69), complex IV (arsenic trioxide) (70) or combined CI/CII inhibitor Mitotane (71) have also been considered for therapy. OXPHOS-directed compounds have been chemically modified for specific delivery into mitochondria and increased efficacy, as demonstrated by mitochondrial-targeted tamoxifen (MitoTam, CI inhibitor) (72), mitochondria-targeted metformin (MitoMet) (73, 74) and mitochondria-targeted αTOS (MitoVES) (75). Several OXPHOS-directed compounds entered clinical trials in cancer. The best developed of these is metformin, which is routinely used for the treatment of diabetes. In addition, IACS-010759 (NCT02882321, NCT03291938) and MitoTam (MitoTam-01 trial; EudraCT 2017-004441-25) are in clinical trials, while a trial with BAY 87-2243 (NCT01297530) has been terminated.
Metformin is the most studied OXPHOS inhibitor in the clinical settings and its effects on the TME are also the best characterized. Metformin was associated with a reduced risk of cancer (60), and a later prospective study (10 years, 1353 patients) showed that metformin in diabetic patients reduces cancer mortality by 43% (76). The related but more potent CI inhibitor phenformin has been withdrawn due to lactic acidosis complications (77, 78). Metformin has pleiotropic effects (CI, adenosine monophosphate-activated protein kinase (AMPK), etc.), but its anti-tumor efficacy has been linked to CI inhibition in cancer cells (79). Interestingly, by inhibiting CI, metformin synergizes with MCT inhibitors to suppress utilization of external lactate (80), which can be produced by CAFs (44, 81).
CAFs promote migration and invasiveness of cancer cells, including those of pheochromocytoma origin (82, 83). Interestingly, metformin treatment of pheochromocytoma/CAFs co-cultures reduced CAF-induced-migration and invasiveness of succinate dehydrogenase subunit B (SDHB)-deficient pheochromocytoma cells grown as spheroids. The underlying mechanism is unclear at present, but likely involves a direct action of metformin on CAFs, as CAFs with diminished mitochondrial oxidative metabolism showed reduced ability to stimulate migration of pheochromocytoma cells (84, 85). Metformin was shown to upregulate AMPK signaling and thus downregulate HIF-1α, transforming growth factor-β (TGF-β) and interleukin 8 (IL-8) in co-culture of CAFs with human breast cancer lines and thus preventing tumor-stroma crosstalk (86). Metformin also decreased production of pro-tumorigenic cytokine IL-6 via NFκB signaling in CAFs cultured with ovarian cancer cell line and upregulated calmodulin−like protein 3 (Calml3) in cultured CAFs that inhibits gastric cancer cell growth (87, 88). Accordingly, metformin may disrupt pro-tumorigenic communication in the TME by hitting both cancer cells and CAFs.
ECs require OXPHOS for angiogenesis and to provide stress resistance in mature vessels (89–91). Multiple OXPHOS-targeting compounds effective against cancer have an anti-angiogenic activity. Complex II inhibitors αTOS and MitoVES (92, 93), as well as arsenic trioxide derivative GSAO (94) inhibit tumor angiogenesis by elevating intracellular ROS which eliminates proliferating ECs. Inhibition of angiogenesis by metformin has been linked to suppression of HIF-1α via AMPK signaling (95, 96), or to inhibition of platelet-derived growth factor β (PDGF-β) that lead to normalization of tumor vasculature (97). Given the pleiotropy of metformin action, it has not been conclusively demonstrated that its anti-angiogenic effect is directly linked to CI inhibition. Still, unrelated, and presumably more specific CI inhibitors BAY 87-2243 (63) and deguelin (98) also inhibit angiogenesis, suggesting that CI inhibition plays a causal role in this effect.
In the immune compartment, OXPHOS supports function of pro-tumorigenic Tregs (99), suggesting that OXPHOS inhibition could improve anti-tumor immune responses. In line, metformin treatment suppresses Tregs in tumors (100) and CI inhibitor MitoTam improves efficacy of immune checkpoint therapy in a pre-clinical renal cancer model (101). Metformin lowers macrophage infiltration in tumors and skews TAM polarization from M2 to anti-tumor M1 phenotype (102, 103). Metformin also protects tumor infiltrating lymphocytes (TILs) from immune exhaustion and apoptosis, supporting tumor rejection in immunocompetent mice, but not in immunodeficient SCID mice (104). Mechanistically, metformin treatment has been shown to promote aberrant glycosylation of programmed death ligand-1 (PD-L1) thus reducing its levels on cancer cells and blocking the PD-L1/PD-1 signaling (105).
Taken together, OXPHOS inhibition may be effective against tumors via direct targeting of cancer cells, inhibition of CAFs, suppression of angiogenesis and by boosting the immune response. However, not all these aspects have been conclusively demonstrated in in vivo tumor models.
Tricarboxylic acid cycle
The TCA cycle (Figure 4) connects cellular metabolism to oxidative phosphorylation and contributes to both catabolism and anabolism. Multiple cancers harbor mutations or deletions in TCA cycle enzymes isocitrate dehydrogenase (IDH), fumarate hydratase (FH), succinate dehydrogenase (SDH) and succinyl-CoA ligase (SUCL) (106–110). As the TCA produces metabolic intermediates with inhibitory effects on enzymes that remove epigenetic marks from the chromatin (6), these alterations affect epigenetics and promote transformation and progression of tumorigenesis.
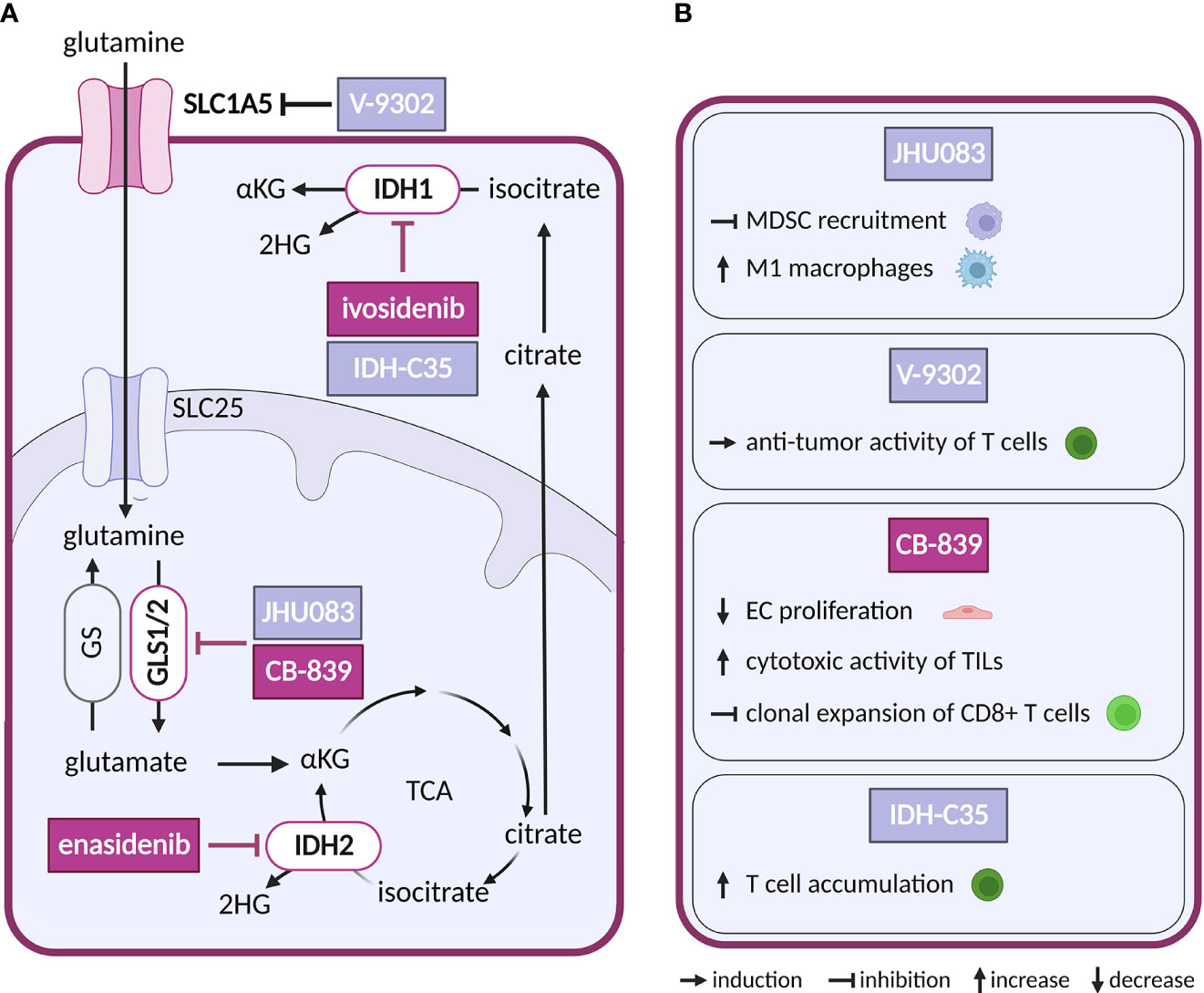
Figure 4 Inhibitors of glutaminolysis or IDH1/2, and their effects on TME cell types. (A) Schematic representation of glutaminolysis and IDH1/2 reactions. Inhibitors of glutamine metabolism with a reported effect in the TME and all inhibitors of IDH1 and IDH2 mentioned in the text are shown. Inhibitors tested in clinical trials for cancer are highlighted in dark violet, while those tested in preclinical studies are shown in light grey-blue. (B) Effects of the glutamine metabolism inhibitors and IDH-C35 shown in (A) on cell populations present in the TME.
So far only IDH1/2-mutated enzymes have been targeted pharmacologically in cancer, while no direct inhibitors are available for FH, SDH or SUCL loss-of-function malignancies. Gain-of-function mutations in isoforms IDH1 and IDH2 (but not in the main TCA isoform IDH3) leading to production of oncometabolite (R)-2-hydroxygultarate (2HG) instead of physiologically produced α-ketoglutarate (αKG) have been found in hematological and solid tumors (111). Increased levels of 2HG inhibit αKG dependent oxygenases which through epigenetic dysregulation promote oncogenesis (112). Small molecule inhibitors of mutant IDH2 and IDH1 are in clinical use in acute myeloid leukemia, including enasidenib (AG-221) and ivosidenib (AG-120). Showing potency both in vitro and in vivo, enasidenib has been in clinical trials in several other types of cancer with mutated IDH2 (NCT02273739, NCT02577406, NCT01915498) (113, 114). With respect to the TME, there are conflicting reports as to the effect of IDH1/2 mutation in cancer cells on the ECs, showing both increased (115, 116) and reduced pro-angiogenic signaling (117). In addition, in intrahepatic cholangiocarcinoma IDH mutations were linked to reduced immune response (118, 119). Reductions in chemokines and suppression of T cell accumulation were reversed by IDH-C35, a specific inhibitor of mutant IDH1 (120). As mutant IDH1/2 are restricted to cancer cells, this effect is very likely secondary to reduction of the oncometabolite. Notably, scRNA-seq analysis of two types of glioblastoma harboring the same IDH1 mutation, astrocytoma and oligodendroglioma, revealed shared developmental hierarchy and common progenitors for all analyzed gliomas, while the TME composition differed between the two tumor types, which is most likely driven by the distinct genetic background (121).
Glutaminolysis
Glutamine, a non-essential AA, is utilized by cancer cells for biomass synthesis and as a source of energy. To meet the increased metabolic needs of rapid proliferation, glutamine is supplied through blood at a concentration as high as 0.5 mM (122). Exogenous glutamine is transported into the cell through solute carrier family 1 member 5 (SLC1A5) but can also be synthesized endogenously by glutamine synthase (GS) in some cell types (Figure 4). Glutamine is further metabolized into glutamate by glutaminases (GLS1/2). Glutamate can be converted to αKG by glutamate dehydrogenase (GLUD) or serve as a nitrogen donor to produce other AAs as well as glutathione for antioxidant defense (123, 124). Via αKG glutamine provides TCA cycle intermediates in a process also known as anaplerosis, which can support proliferation in nutrient-restricted environments (123, 124).
Given the ubiquitous role of glutamine as a substrate, it is not surprising that glutamine utilizing reactions are targeted for anticancer treatment. Mimetic compounds, such as 6-diazo-5-oxo-L-norleucine (DON), bind covalently to the active sites of multiple glutamine-dependent enzymes. DON and other glutamine mimetics, acivicin and azaserine, were explored as potential anticancer treatment (125), but their success is limited by high toxicity. To avoid this toxicity, prodrugs of DON were synthetized to release the active payload at specific sites (126). The prodrug DRP-104 (sirpiglenastat) is being tested in an ongoing clinical trial in advanced stage solid tumors as a single treatment and in combination with anti-PD-L1 antibody (atezolizumab) (NCT04471415). Blocking of glutamine metabolism using DON derived prodrug, JHU083, in the TME has not only directly impacted cancer cells, but also acted on the immune compartment. It inhibited recruitment of myeloid derived suppressor cells (MDSCs) and increased generation of antitumor inflammatory TAMs, leading to decreased metastatic potential and improved anti-tumor immunity (127, 128).
In glutamine deprived conditions within the TME, CAFs can increase glutamine synthesis to provide glutamine to cancer cells (129). Thus, targeting glutamine uptake into cancer cells by inhibitors of SLC1A5, such as V-9302, would interrupt such communication. This approach has been explored experimentally in preclinical studies with triple negative breast cancer. Intriguingly, V-9302 suppressed glutamine uptake in cancer cells but not in CD8+ T cells, as T cells were able to upregulate an alternative glutamine transporter. Hence, glutamine availability for T cells increased, stimulating glutathione synthesis in T cells and improving their antioxidant status and anti-tumor activity (130, 131).
As an alternative to glutamine mimetic compounds, researchers have targeted the first enzyme of the glutaminolysis pathway, GLS, which is frequently upregulated in cancer (132). Several allosteric inhibitors have been designed to target isoforms of GLS, such as CB-839 (telaglenastat) and bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)-ethyl sulfide (BPTES) (133, 134). CB-839 has been tested both as a single treatment and in combination with antimetabolites and immunotherapy (azacytidine, nivolumab), in several types of cancer (NCT02071927, NCT02771626). For example, a combination with glutamine uptake inhibitor V-9302 overcame resistance of hepatocellular carcinoma, a glutamine dependent cancer type otherwise resistant to CB-839 alone (135). Besides the direct effect on cancer cells, CB-839 improved the cytotoxic activity of TILs. By altering glutamine metabolism in cancer cells, CB-839 increased extracellular glutamine levels available to T cells for GLS independent activities such as antigen activation (136). On the other hand, in lung adenocarcinoma CB-839 had an opposite effect on CD8+ T cells, inhibiting their clonal expansion and activation induced by immunotherapy (137). GLS may also be an interesting target in ECs, where it supports proliferation that is reduced by CB-839 treatment (138).
BPTES has been used for targeting KRAS-mutated cancer cells that are addicted to glutamine. Administration of BPTES inhibited tumor growth in Burkitt lymphoma model, through metabolic alteration of glutamate, αKG, succinate and fumarate while additionally increasing ROS levels (139). Consistent with GLS1 supporting pathological angiogenesis, impairment of angiogenesis was observed in ECs treated with a combination of BPTES and a PDK inhibitor DCA (22, 138). BPTES also upregulated PD-L1 on cancer cells, which interfered with T cell activity. This was mitigated by co-treatment with anti-PD-L1 antibody, which rescued T cell function (140). In recent years there has been effort to utilize more specific approaches to BPTES delivery using nanoparticles to avoid systemic and off target effects (141).
GS has been shown as critical for EC motility and migration, processes that underlie angiogenesis during development and in pathological states, making GS an attractive target for inhibition of pathological angiogenesis (142). Furthermore, genetic deletion of GS in TAMs decreases glutamine in TME, which results in tumor vessel normalization, reactivated T cells and decreased cancer cell motility (143).
Overall, these results suggest that more specific targeting and combination therapy might be necessary to overcome systemic toxic effect of glutamine deprivation therapy as well as complex metabolic crosstalk present in TME.
Amino acids
Metabolism of proliferating cancer cells may become dependent also on other AAs that are normally non-essential, providing therapeutic opportunities in reducing AA serum levels (144). The first AA targeted in this manner was asparagine. An enzyme called L-asparaginase (Ciderolase, Erwinase), originally identified in guinea pig serum (145, 146), can degrade blood asparagine and hereby limit its availability. L-asparaginase is effective because the enzyme asparagine synthase (ASNS, responsible for endogenous asparagine synthesis) is decreased in some cancer cells, making them dependent on exogenous asparagine for proliferation (147, 148). At present, L-asparaginase is an approved therapy in ALL and non-Hodgkin lymphoma, while in other leukemias and solid cancers it is in clinical trials (NCT03665441, NCT01574274). Beside cancer cells, L-asparaginase treatment may also affect other cell types in the TME. ECs utilize asparagine for sprouting and vessel growth, particularly under glutamine deprivation (138), while CD8+ T cells need extracellular asparagine for optimal immune response (149), hinting at a possible complex response to asparagine depletion in the TME.
Arginine is synthetized through subsequent reaction of argininosuccinate synthase (ASS) and argininosuccinate lyase (ASL). Interestingly, a subset of malignancies do not express ASS, opening a potential therapeutic window for arginine deprivation therapy using arginine-degrading enzymes arginine deiminase (ADI) (150, 151) or arginase (152). ADI has progressed into phase 3 trials in cancer, both as a single agent and a combinatorial therapy (NCT02709512, NCT05317819). In the TME arginine depletion impaired T cell responses. Chimeric antigen T cells (CAR-T) proliferation was rescued by expression of the ASS and ornithine transcarbamylase (OTC) without loss of cytotoxicity of CAR-Ts, suggesting that reengineered cells prevail in arginine depleted environment (153). In addition, extracellular arginine shifts T cell metabolism from glycolysis to oxidative phosphorylation, priming them for activation and generation of memory cells (154). To locally increase arginine availability to T cells, an E. Coli strain was engineered that converts ammonia to arginine for enhanced efficacy of immunotherapy. Arginine level in tumor homogenates was increased following intratumoral injection of engineered E. Coli, the number of TILs was elevated and there was a synergistic effect with PD-L1 blocking therapy (155).
Methionine is an essential AA, still the dependence of cancer cell on methionine is particularly pronounced (156). Unlike normal cells, proliferating cancer cell are unable to utilize/recycle methionine precursor homocysteine (157). Several approaches were developed to decrease serum levels of methionine, including the enzyme methionase and dietary restrictions (158). While depleting methionine from serum proved challenging to strike the balance between toxicity and benefits, recently there was a reemergence of this approach (159, 160). With respect to TME, in hepatocellular carcinoma patients increased excretion of methionine metabolism intermediates from cancer cells induced T cell exhaustion due to epigenetic changes, suggesting that reducing methionine availability to cancer cells could stimulate immune response (159). On the other hand, low levels of methionine in the TME, due to excessive consumption by cancer cells, also reduced anti-tumor immunity via epigenetic changes in T cells (160). Thus, selective inhibition of methionine uptake by cancer cells but not immune cells, similarly as previously shown for glutamine (130), might be the most effective way to preserve and boost anti-tumor immunity.
Proline is a non-essential amino acid. Nevertheless, proline synthesis enzymes are frequently upregulated in cancer (161, 162) and endogenous proline synthesis supports cancer cell proliferation. While exogenous proline can be obtained from the TME, de novo proline synthesis in cancer cells is required to maintain cellular redox state via NADH recycling by 5-carboxylate reductase 1 (PYCR1) (163). On the other hand, proline degradation by proline dehydrogenase (PRODH) sustains breast cancer metastases and promotes epithelial to mesenchymal transition in lung cancer, effects sensitive to PRODH inhibitor L-tetrahydro-2-furoic acid (L-THFA) (164, 165). Proline synthesis, both in cancer cells and in CAFs, modulates the TME. PYCR1 in cancer cells in conjunction with the mitochondria protease Lon elicits ROS-dependent production of pro-inflammatory cytokines that promote M2 macrophage polarization and angiogenesis (165). Proline synthesis in CAFs then enables production and deposition of collagen, a major component of the extracellular matrix. Suppression of PYCR1 in CAFs inhibits collagen deposition, tumor growth and metastatic potential in breast cancer (3). Consistently, autophagy deficiency in CAFs impedes proline biosynthesis and collagen deposition (166). Inhibitors of proline metabolism have not yet been evaluated in clinical trials. However, PYCR1 expression in CAFs is epigenetically regulated and depends on cytosolic acetyl-CoA (3). Besides PYCR1 inhibitors that are under development (167), more established agents that interfere with acetyl-CoA management (inhibitors of histone acetyl transferase EP300 or ATP citrate lyase) could be considered for therapy.
Fatty acid metabolism
Fatty acid (FA) metabolism (Figure 5) is important for proliferation and survival of cancer cells under conditions of glucose limitation, where FA β-oxidation (FAO) serves as an alternative energy source (168). Indeed, upregulation of a key enzyme in FAO, carnitine O-palmitoyltransferase 1 (CPT1) protects cancer cells in glucose-deprived conditions and CPT1 knockdown sensitizes cells to therapy (169). Conversely, overexpression of lipogenic enzymes such as acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN) is commonly seen in tumors and linked to poor prognosis (168).
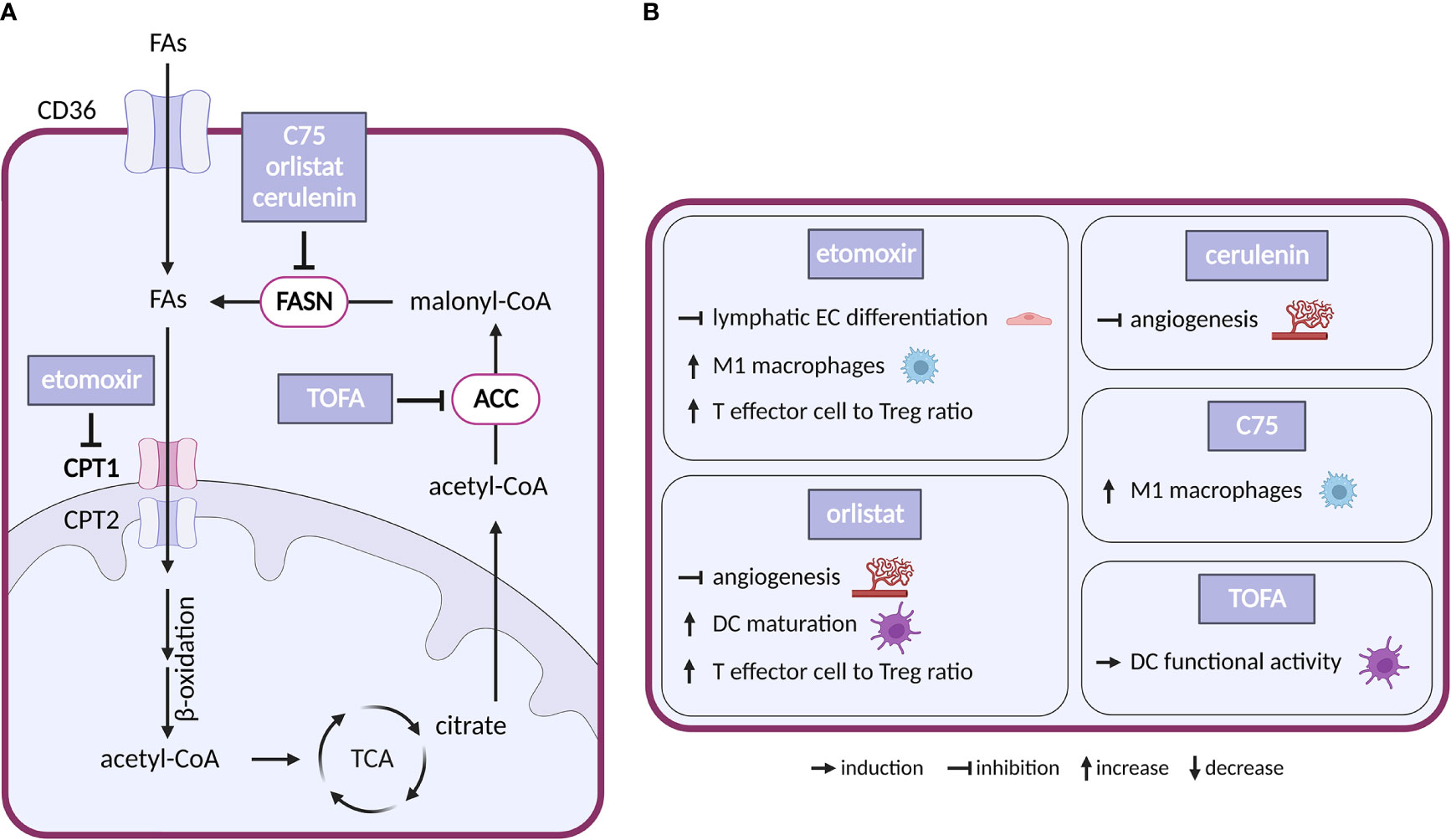
Figure 5 Inhibitors of fatty acid metabolism and their effects on TME cell types. (A) Schematic representation of FA metabolism. Only inhibitors with a reported effect in the TME are shown. Note that none of these inhibitors entered clinical trial in cancer. (B) Effect of the inhibitors shown in (A) on cell populations present in the TME.
Inhibitors of enzymes of lipid metabolism (FASN, ACC, CPT1) have been intensively studied and several compounds have shown efficacy in experimental settings: (i) With respect to FASN inhibitors, orlistat, a clinically used anti-obesity drug (a pancreatic lipase blocker), shows anticancer effectivity in multiple cancer models (170). Cerulenin and its synthetic analogue C75 slowed down the growth of breast and ovarian cancer xenografts (171, 172). Besides that, number of new-generation FASN inhibitors have been developed recently including TVB-2640, which is now in clinical trials in cancer (NCT02223247). (ii) Regarding ACC inhibitors, 5−tetradecyloxy−2−furoic acid (TOFA) inhibited the growth of ovarian tumor xenograft via downregulation of cell-cycle regulating proteins (173). (iii) For CPT1 inhibitors, etomoxir decreased growth of prostate cancer and leukemia xenografts in mice (174, 175). However, the drug has been discontinued in phase II clinical trial for treatment of heart failure due to hepatotoxicity (176). Another CPT1 inhibitor, ST1326, prevented formation of B-cell lymphoma in mice (177) and CPT1 inhibitor ranolazine administered together with pyruvate dehydrogenase kinase inhibitor DCA inhibited growth of glioblastoma in mice (178).
In the TME, CAFs often feature elevated lipid biosynthesis and upregulation of FASN expression, which promotes lipid transfer from CAFs to cancer cells and stimulates tumor growth (179, 180). Silencing of FASN in CAFs reduced migration of colorectal cancer cells grown in CAF-conditioned media (181). Interestingly, CPT1 was downregulated in metastatic colorectal cancer, but upregulated in the corresponding CAFs. Consistently, in mice DLD1 colorectal cancer cells formed bigger tumors when injected with CAFs overexpressing CPT1A compared to normal CAFs. It appears that in this arrangement the preferential use of FAs by CAFs saves glucose for cancer cells. Pharmacological inhibition of overexpressed CPT1A with etomoxir resulted in reduced tumor growth (182).
Vascular ECs consume FAs as a carbon source for DNA (but not RNA) synthesis. Loss of CPT1 in vascular ECs impairs angiogenesis (183) because of a proliferation (but not migration) defect. CPT1 loss in lymphatic ECs alters epigenetics due to reduced coenzyme A levels and impairs lymphangiogenesis (184). In the adult vasculature, FAO supports vascular integrity (185, 186). Similarly, disruption of FA synthesis (by FASN deficiency) results in defects in angiogenesis and increased vessel permeability linked to decreased palmitoylation, reduced activity of endothelial nitric-oxide synthase, increased malonylation and reduced activity of the mTORC complex 1 (mTORC1) (187). Pharmacological modulation of FA metabolism in ECs recapitulated these effects. CPT1 blockade with etomoxir inhibited lymphatic EC development and differentiation (184). FASN inhibitors orlistat and cerulenin inhibited angiogenesis and slowed down the growth of B16-F10 melanoma tumors in mice (188, 189).
In the immune compartment, FA metabolism regulates differentiation of TAMs. FAO is upregulated in alternatively-activated macrophages that tend to be immunosuppressive (190). FAs, and in particular polysaturated FAs, are abundant in TME, and palmitate or oleate treatment stimulates transition of macrophages towards the immunosuppressive M2 phenotype (191–193). This effect is abolished by the CPT1 inhibitor etomoxir (193), which also promotes the pro-inflammatory M1 phenotype (194). Similarly, linoleic acid, via lipid-sensing peroxisome proliferator–activated receptor (PPAR) β/δ signaling, imparts TAMs with pro-tumorigenic properties (195). Interestingly, the FASN inhibitor C75 has inhibited lipid droplet formation and stimulated M1 phenotype via MEK1/2 axis in Raw264.7 macrophage cell line (194). These results indicate that the FAs of the TME shape TAM’s phenotype and suggest that pharmacological targeting of FAs could be used to promote anti-tumor immunity.
FA metabolism can also determine the fate of T cells and change their repertoire. While CD8+ effector T cells restrict tumorigenesis by attacking cancer cells, Tregs promote tumorigenesis by suppressing the effector T cells (196). Free FAs are toxic for effector T cells in high concentration, but they promote Tregs whose metabolism favors FA oxidation (197). Thus, while generally TME might accentuate Tregs over T effectors, the inhibition of FA biosynthesis might have opposing effects. Indeed, breast cancer cells release oleate which compromises T effectors (198), and etomoxir treatment resulted in slower growth of glioblastoma in mice and increasing T effector to Treg ratio (199). Interestingly, inhibition of FAO with etomoxir or perhexiline enhanced glycolysis in T effectors in obesity-linked breast cancer, which resulted in reduced breast mammary tumor growth in obese mice (200). These results are further confirmed by a recent study of PPARγ signaling in T effectors, where PPARγ agonist bezafibrate induced mitochondrial respiration and FAO, which improved survival of effector T cells in FA rich TME (201).
Unlike for most other metabolic treatments, the effects of inhibiting FA metabolism were described in dendritic cells (DCs). DCs are important for establishing the immune response as they present antigens to T cells. Lipid accumulation resulted in dysfunction of DCs in antigen presentation (196). Inhibiting FA biosynthesis with ACC inhibitor TOFA suppressed melanoma tumor growth in mice and improved ability of DCs to stimulate T cells (202). Complex recent study showed that inhibition of FA biosynthesis with FASN inhibitor orlistat also resulted in higher T effector and reduced Treg tumor infiltration, increased DC maturation and slowed down tumor growth (203). FAs could potentially also dampen the innate anti-tumor immune response, namely its most important effectors, the NK cells. In lipid rich TME, NK cells tend to accumulate and store lipids to prevent lipid toxicity, but when this intracellular lipid accumulation becomes excessive, it can disrupt proper NK cell function (196).
Nucleotides
In highly proliferating cells, purine and pyrimidine nucleotides are indispensable for nucleic acid synthesis. De novo synthesis of nucleotides (Figure 6) requires input from pentose phosphate pathway, TCA cycle, OXPHOS and one-carbon pool (204). Energetically less demanding salvage pathways, that recycle intracellular or extracellular nucleoside pools, are generally preferred in differentiated cells (205), although preferential use of salvage pathways has also been observed in breast cancer during epithelial–mesenchymal transition (206).
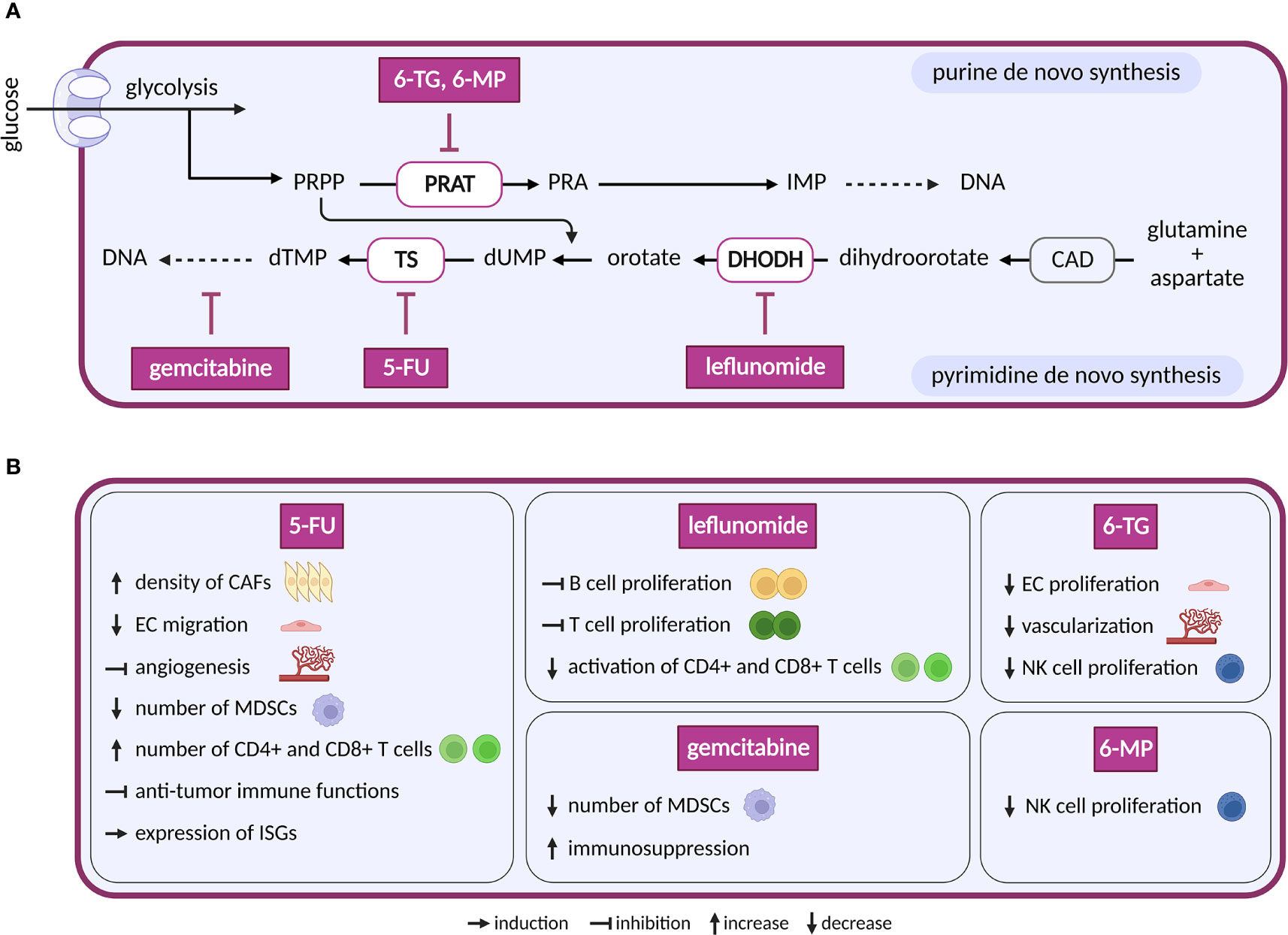
Figure 6 Inhibitors of nucleotide metabolism and their effects on TME cell types. (A) Schematic representation of nucleotide de novo synthesis. Anti-nucleotide therapy compounds with a reported effect in the TME are shown. These compounds are in clinical trials for cancer or in clinical use. (B) Diverse effects of the nucleotide inhibitors shown in (A) on cell populations present in the TME.
The first drug to target de novo nucleotide synthesis, aminopterin, was successful in patients with acute lymphoblastic leukemia. Aminopterin is a folate analog that inhibits dihydrofolate reductase-dependent synthesis of tetrahydrofolate (207). Tetrahydrofolate is essential in the production of purines and pyrimidines, and its deficiency results in lower DNA, RNA and protein synthesis. Newer derivates of aminopterin, such as methotrexate, trimetrexate and pemetrexed, are called by a common name antifolates (208). The initial clinical success of antifolates led to the development of a new class of drugs called antimetabolites. Antimetabolites are analogues of purines and pyrimidines that interfere with proliferation in two different ways: by inhibition of enzymes involved in nucleotide base synthesis and by incorporation into DNA or RNA (209, 210). Several antimetabolites are currently approved for cancer treatment. The purine analogs 6-mercaptopurine (6-MP) and 6-thioguanine (6-TG) inhibit phosphoribosyl pyrophosphate amidotransferase, the first enzyme in de novo purine biosynthesis (209). The pyrimidine analog 5-fluorouracil (5-FU) and its prodrug capecitabine inhibit thymidylate synthase and therefore block synthesis of dTTP needed for DNA replication. Cytarabine and gemcitabine are analogues of pyrimidine deoxycytidine. Both incorporate into the DNA and stop DNA replication (211).
Compounds that directly target de novo synthesis of pyrimidine are also available. The most druggable enzyme of pyrimidine synthesis is dihydroorotate dehydrogenase (DHODH) (212), and multiple DHODH inhibitors have been designed, including brequinar, leflunomide, teriflunomide, ASLAN003, BAY2402234 and several others (213). While effective in preclinical mouse models of small cell lung cancer (214), breast cancer, pancreatic cancer and glioblastoma (215, 216), clinical trials in human cancer were less successful. Possibly, cancer cells may overcome nucleotide shortage via salvage pathway (217). However, dipyridamole, an inhibitor of nucleoside transport via solute carrier family 29 member 1 (SLC29A1/ENT1), showed synergistic effect with brequinar in vitro, but not in vivo (218). This might be due to rescue by uridine in plasma that can rebound after the use of brequinar (219).
Nucleotide-based therapy has satisfactory outcomes, but its general anti-proliferative effects lack selectivity. Consequently, it also affects cells of bone marrow, intestine and hair follicles, which leads to toxicity (220). Many cells of the TME also have a proliferative character and depend on nucleotides for DNA/RNA synthesis. TECs, for instance, enhance nucleotide synthesis and upregulate several enzymes required for purine and pyrimidine synthesis (20, 221). 5-FU and 6-TG decrease migration and/or proliferation of primary ECs and in vivo angiogenesis. Inhibition of pyrimidine synthesis by 5-FU affects migration capacity of ECs stimulated by VEGF and FGF-2, tube formation stimulated by VEGF (222) and angiogenesis induced by murine renal cell carcinoma in dorsal air sac assay (223). Similarly, purine analogue 6-TG inhibited fetal bovine aortic EC proliferation upon VEGF and FGF-2 stimulation. Consistently, increased vascularization of bone marrow in patients with AML was reverted under 6-TG treatment (224) and inhibition of pathways feeding into nucleotide synthesis in ECs such as FAO (183), glutaminolysis (138) and glycolysis (20) diminished proliferation. These effects on ECs might lead to decreased vascularization and subsequently to slower tumor growth.
Compared to ECs, CAFs are less well studied regarding consequences of nucleotide synthesis inhibition. Interestingly, increased density of CAFs was observed following treatment with maximum tolerated dose of 5-FU in vivo (225) and in patients with combined treatment of 5-FU and radiotherapy (226).
In the immune compartment, a negative effect on patient immune response is expected, as activated T and B cells metabolically depend on nucleotide synthesis for their proliferation (227–229). Leflunomide, the most clinically developed inhibitor of pyrimidine de novo synthesis, blocks proliferation of antibody producing B cells (229), malignant B cells (230), T cells (231) and interferes with activation of CD4+ T and CD8+ T cells in HIV-1 infected patients (232). In a murine autoimmune disease model, leflunomide suppressed B and T cells. Uridine supplementation was able to rescue activity of suppressed cells with the exception of CD8+ T cells (233). As supplementation with exogenous uridine normally compensates for defects/inactivity of de novo pyrimidine synthesis, this points to possible off target effects of leflunomide, such as inhibition of MAPK or tyrosine kinases (234). Hence, newer, and more specific inhibitors of DHODH might provide better outcomes. Interestingly, one dose treatment 5-FU reduced the number of MDSCs and therefore activated immune response by stimulating CD8+ T cells (235). Additionally, 5-FU treatment elevated the number of tumor-associated CD4+ and CD8+ T cells (236). However, another study documented impaired anti-tumor immune functions with more 5-FU treatment cycles (237). Similar to pyrimidines, inhibitors of purine metabolism also negatively impact immune cells. 6-TG and 6-MP decreased proliferation of NK cells from peripheral blood (238). Elevated purine levels in proliferating cells increase expression of MHC class I polypeptide-related sequence and in this way stimulate NK group 2D effectors to recognize abnormal cells (239). Notably, antimetabolites are also used as immunosuppressants (240), underscoring their potential to dampen immune response. Surprisingly therefore, they can also support protective immunity. Similar to 5-FU, one-time treatment of mice bearing large tumors with gemcitabine, an analog of pyrimidine deoxycytidine, depleted MDSCs in the spleen (241, 242). However, repeated dose of gemcitabine promoted immunosuppression in the TME (243). Additionally, in an in vitro model of viral infection (243) and mouse embryonic fibroblasts (244), inhibition of de novo pyrimidine synthesis induced expression of interferon-stimulated genes (ISGs) that are known to promote innate immune response. Current evidence points to predominantly suppressed immune response upon inhibition of nucleotide synthesis. However, these negative effects on immune response can be exploited for therapy. For example, pemetrexed promotes immune checkpoint blockade through per se undesired transcriptional activation of PD-L1 in cancer cells, and activation of T-lymphocytes was observed in vitro when pemetrexed was combined with the anti-PD-1/PD-L1 therapy (245).
Conclusion
Metabolic adaptations that promote proliferation and survival of tumor cells represent an attractive target for cancer therapy. However, such metabolic therapy is challenged by the intrinsic flexibility and heterogeneity of cancer metabolism, the narrow therapeutic window due to an overlap with the metabolism of healthy cells (246), and by the complex interactions of metabolic agents in the TME. The available information about how TME responds to metabolic treatments is often limited, indirect and derived from sub-optimal experimental models. The emerging patterns nevertheless suggest that metabolic treatments could substantially impact the TME both to enhance or reduce the efficacy of the intended therapeutic interventions. Although we consider it unlikely that unfavorable interactions in the TME would lead to an outright acceleration of primary tumor growth, they might reduce effectiveness, prevent a lasting remission, and perhaps increase the risk of metastasis.
With respect to the major TME cell types, the immune compartment seems to be the most affected, and is often impacted in a negative manner. Immune cells, particularly effector T cells, need to expand rapidly when immune response is activated. Treatments that target rapidly proliferating cancer cells can therefore reduce the numbers and potency of anti-tumor effectors. Potential approaches to inhibit glycolysis, nucleotide metabolism, glutamine or arginine should thus be carefully evaluated for their impact on T cells. In contrast, therapies that limit Tregs (targeting OXPHOS and FA metabolism mostly) have a very good potential to improve outcomes of therapy.
Like immune cells, ECs also proliferate during tumor angiogenesis and rely mostly on glycolysis. An outright elimination of TECs is not warranted, as this impairs vessel function, promotes tumor hypoxia and can potentially induce metastases. On the other hand, partial inhibition of pro-angiogenic metabolic pathways such as glycolysis that reduces EC proliferation and angiogenic activity, leads to vessel normalization, increased perfusion and better response to combinatorial therapy.
Targeting CAFs with current metabolic treatment seem to have the least negative effects, as there is little evidence that CAFs would decrease effectiveness of existing metabolic drugs. Instead, these drugs tend to reduce nutrients and the overall support that CAFs provide to cancer cells. Proline synthesis seems to be a particularly promising target in CAFs, yet at present there are only limited options for pharmacological targeting of this pathway.
What is the best way forward? We see considerable potential in combinatorial metabolic therapy. Agents targeting OXPHOS and FA metabolism seem to show the most benefits in the TME and may thus be good candidates for such combinations. Furthermore, metabolic drugs often target fundamental metabolic processes that are shared across cell types. It might therefore be convenient to focus on auxiliary components of these pathways such as plasma membrane transporters that are more cell type specific, allowing differentiation between cancer cells and for example T cells. A prime example of such strategy is the glutamine uptake inhibitor V-9302, in response to which effector T cells, but not cancer cells, upregulate an alternative transporter making them insensitive. In the long run, future treatments would benefit from design of compounds that are less easily taken up by certain TME cell types such as effector T cells. Similarly, engineered CAR-T cells could in principle be designed as resistant to metabolic inhibitors.
To sum up, when carefully considered, finetuning the effects of metabolic treatments in the TME represents a promising opportunity for new metabolic anti-cancer strategies.
Author contributions
PH prepared figures. All authors prepared and revised the manuscript and approved the final version.
Acknowledgments
The authors apologize to any author whose work could not be included due to space limitation. All figures were created with BioRender.com.
Funding
MM is supported by the Grant Agency of Charles University (1435320). The work of JR is supported by the Czech Science Foundation (20-18513S, 22-34507S) and Czech Health Research Council (NU22-07-00087). The work of KR is supported by a Marie Skłodowska-Curie IF (101027977), by EMBO Installation Grant (5068-2022) and by an ERC Starting Grant (101042031). Institute of Biotechnology receives infrastructural support from the Czech Academy of Sciences (RVO86652036).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Fendt SM. Is there a therapeutic window for metabolism-based cancer therapies? Front Endocrinol (Lausanne) (2017) 8:150. doi: 10.3389/fendo.2017.00150
2. Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front Cell Dev Biol (2019) 7:60. doi: 10.3389/fcell.2019.00060
3. Kay EJ, Paterson K, Riera-Domingo C, Sumpton D, Dabritz JHM, Tardito S, et al. Cancer-associated fibroblasts require proline synthesis by Pycr1 for the deposition of pro-tumorigenic extracellular matrix. Nat Metab (2022) 4(6):693–710. doi: 10.1038/s42255-022-00582-0
4. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the tca cycle Via circulating lactate. Nature (2017) 551(7678):115–8. doi: 10.1038/nature24057
5. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
6. Pavlova N, Thompson C. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
7. Lunt SY, Vander Heiden MG. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27(1):441–64. doi: 10.1146/annurev-cellbio-092910-154237
8. Li J, Eu JQ, Kong LR, Wang L, Lim YC, Goh BC, et al. Targeting metabolism in cancer cells and the tumour microenvironment for cancer therapy. Molecules (2020) 25(20):4831. doi: 10.3390/molecules25204831
9. Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of atp-producing pathways. Science (2001) 292(5516):504–7. doi: 10.1126/science.1058079
10. Landau BR, Laszlo J, Stengle J, Burk D. Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-Deoxy-D-Glucose. JNCI: J Natl Cancer Inst (1958) 21(3):485–94. doi: 10.1093/jnci/21.3.485
11. Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol Res (2019) 150:104511. doi: 10.1016/j.phrs.2019.104511
12. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer-associated fibroblasts by Idh3α downregulation. Cell Rep (2015) 10(8):1335–48. doi: 10.1016/j.celrep.2015.02.006
13. Becker LM, O'Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep (2020) 31(9):107701. doi: 10.1016/j.celrep.2020.107701
14. Fitzgerald G, Soro-Arnaiz I, De Bock K. The warburg effect in endothelial cells and its potential as an anti-angiogenic target in cancer. Front Cell Dev Biol (2018) 6:100. doi: 10.3389/fcell.2018.00100
15. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of Pfkfb3-driven glycolysis in vessel sprouting. Cell (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
16. Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, et al. Fgf-dependent metabolic control of vascular development. Nature (2017) 545(7653):224–8. doi: 10.1038/nature22322
17. Yetkin-Arik B, Vogels IMC, Nowak-Sliwinska P, Weiss A, Houtkooper RH, Van Noorden CJF, et al. The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci Rep (2019) 9(1):12608. doi: 10.1038/s41598-019-48676-2
18. Merchan JR, Kovács K, Railsback JW, Kurtoglu M, Jing Y, Piña Y, et al. Antiangiogenic activity of 2-Deoxy-D-Glucose. PloS One (2010) 5(10):e13699. doi: 10.1371/journal.pone.0013699
19. Del Bufalo D, Trisciuoglio D, Scarsella M, D'Amati G, Candiloro A, Iervolino A, et al. Lonidamine causes inhibition of angiogenesis-related endothelial cell functions. Neoplasia (2004) 6(5):513–22. doi: 10.1593/neo.04133
20. Cantelmo AR, Conradi L-C, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the glycolytic activator Pfkfb3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell (2016) 30(6):968–85. doi: 10.1016/j.ccell.2016.10.006
21. Conradi L-C, Brajic A, Cantelmo AR, Bouché A, Kalucka J, Pircher A, et al. Tumor vessel disintegration by maximum tolerable Pfkfb3 blockade. Angiogenesis (2017) 20(4):599–613. doi: 10.1007/s10456-017-9573-6
22. Schoonjans CA, Mathieu B, Joudiou N, Zampieri LX, Brusa D, Sonveaux P, et al. Targeting endothelial cell metabolism by inhibition of pyruvate dehydrogenase kinase and glutaminase-1. J Clin Med (2020) 9(10):3308. doi: 10.3390/jcm9103308
23. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early tcr signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep (2018) 22(6):1509–21. doi: 10.1016/j.celrep.2018.01.040
24. Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. Mtorc1-dependent metabolic reprogramming is a prerequisite for nk cell effector function. J Immunol (2014) 193(9):4477–84. doi: 10.4049/jimmunol.1401558
25. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
26. Pavlou S, Wang L, Xu H, Chen M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J Inflammation (2017) 14(1):4. doi: 10.1186/s12950-017-0151-x
27. Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in Cd8 versus Cd4 T cells. PloS One (2014) 9(8):e104104. doi: 10.1371/journal.pone.0104104
28. Martins CP, New LA, O'Connor EC, Previte DM, Cargill KR, Tse IL, et al. Glycolysis inhibition induces functional and metabolic exhaustion of Cd4(+) T cells in type 1 diabetes. Front Immunol (2021) 12:669456. doi: 10.3389/fimmu.2021.669456
29. Ho P-C, Dauz Bihuniak J, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
30. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
31. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. Hif1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of Th17 and treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
32. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. Tlr8-mediated metabolic control of human treg function: A mechanistic target for cancer immunotherapy. Cell Metab (2019) 29(1):103–23.e5. doi: 10.1016/j.cmet.2018.09.020
33. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances Cd8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/jci69589
34. Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest (2008) 118(12):3930–42. doi: 10.1172/JCI36843
35. Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab (2020) 33:48–66. doi: 10.1016/j.molmet.2019.07.006
36. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest (2013) 123(9):3685–92. doi: 10.1172/jci69741
37. Corbet C, Bastien E, Draoui N, Doix B, Mignion L, Jordan BF, et al. Interruption of lactate uptake by inhibiting mitochondrial pyruvate transport unravels direct antitumor and radiosensitizing effects. Nat Commun (2018) 9(1):1208. doi: 10.1038/s41467-018-03525-0
38. Roy A, Bera S. Caf cellular glycolysis: Linking cancer cells with the microenvironment. Tumour Biol (2016) 37(7):8503–14. doi: 10.1007/s13277-016-5049-3
39. Domingo-Vidal M, Whitaker-Menezes D, Mollaee M, Lin Z, Tuluc M, Philp N, et al. Monocarboxylate transporter 4 in cancer-associated fibroblasts is a driver of aggressiveness in aerodigestive tract cancers. Front Oncol (2022) 12:906494. doi: 10.3389/fonc.2022.906494
40. Sonveaux P, Copetti T, De Saedeleer CJ, Végran F, Verrax J, Kennedy KM, et al. Targeting the lactate transporter Mct1 in endothelial cells inhibits lactate-induced hif-1 activation and tumor angiogenesis. PloS One (2012) 7(3):e33418. doi: 10.1371/journal.pone.0033418
41. Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, et al. Inhibition of monocarboxylate transporter-1 (Mct1) by Azd3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther (2014) 13(12):2805–16. doi: 10.1158/1535-7163.mct-13-1091
42. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. Ldha-associated lactic acid production blunts tumor immunosurveillance by T and nk cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
43. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol (2015) 11(1):9–15. doi: 10.1038/nchembio.1712
44. Li F, Simon MC. Cancer cells don't live alone: Metabolic communication within tumor microenvironments. Dev Cell (2020) 54(2):183–95. doi: 10.1016/j.devcel.2020.06.018
45. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
46. Zhang L, Li S. Lactic acid promotes macrophage polarization through mct-Hif1α signaling in gastric cancer. Exp Cell Res (2020) 388(2):111846. doi: 10.1016/j.yexcr.2020.111846
47. Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, et al. Monocarboxylate transporter Mct1 is a target for immunosuppression. Nat Chem Biol (2005) 1(7):371–6. doi: 10.1038/nchembio744
48. Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab (2019) 29(2):399–416.e10. doi: 10.1016/j.cmet.2018.10.014
49. Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol (2018) 20(7):775–81. doi: 10.1038/s41556-018-0118-z
50. Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, et al. Asparagine couples mitochondrial respiration to Atf4 activity and tumor growth. Cell Metab (2021) 33(5):1013–26 e6. doi: 10.1016/j.cmet.2021.02.001
51. Sullivan B,L, Gui Y,D, Hosios M,A, Bush N,L, Freinkman E,H, Matthew V,G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. . Cell (2015) 162(3):552–63. doi: 10.1016/j.cell.2015.07.017
52. Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci (2011) 89(3-4):65–71. doi: 10.1016/j.lfs.2011.05.010
53. Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab (2015) 21(1):81–94. doi: 10.1016/j.cmet.2014.12.003
54. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature (2014) 514(7524):628–32. doi: 10.1038/nature13611
55. Reznik E, Miller ML, Senbabaoglu Y, Riaz N, Sarungbam J, Tickoo SK, et al. Mitochondrial DNA copy number variation across human cancers. Elife (2016) 20(11):2666–77. doi: 10.7554/eLife.10769
56. Vaupel P, Mayer A. Availability, not respiratory capacity governs oxygen consumption of solid tumors. Int J Biochem Cell Biol (2012) 44(9):1477–81. doi: 10.1016/j.biocel.2012.05.019
57. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell (2016) 164(4):681–94. doi: 10.1016/j.cell.2015.12.034
58. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res (2018) 24(11):2482–90. doi: 10.1158/1078-0432.CCR-17-3070
59. Farhadi P, Yarani R, Dokaneheifard S, Mansouri K. The emerging role of targeting cancer metabolism for cancer therapy. Tumour Biol (2020) 42(10):1010428320965284. doi: 10.1177/1010428320965284
60. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ (2005) 330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7
61. Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, et al. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer (2012) 106(6):1117–22. doi: 10.1038/bjc.2012.56
62. Varughese RS, Lam WS, Marican A, Viganeshwari SH, Bhave AS, Syn NL, et al. Biopharmacological considerations for accelerating drug development of deguelin, a rotenoid with potent chemotherapeutic and chemopreventive potential. Cancer (2019) 125(11):1789–98. doi: 10.1002/cncr.32069
63. Ellinghaus P, Heisler I, Unterschemmann K, Haerter M, Beck H, Greschat S, et al. Bay 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med (2013) 2(5):611–24. doi: 10.1002/cam4.112
64. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24(7):1036–46. doi: 10.1038/s41591-018-0052-4
65. Moreira PI, Custodio J, Moreno A, Oliveira CR, Santos MS. Tamoxifen and estradiol interact with the flavin mononucleotide site of complex I leading to mitochondrial failure. J Biol Chem (2006) 281(15):10143–52. doi: 10.1074/jbc.M510249200
66. Guo L, Shestov AA, Worth AJ, Nath K, Nelson DS, Leeper DB, et al. Inhibition of mitochondrial complex ii by the anticancer agent lonidamine. J Biol Chem (2016) 291(1):42–57. doi: 10.1074/jbc.M115.697516
67. Dong LF, Freeman R, Liu J, Zobalova R, Marin-Hernandez A, Stantic M, et al. Suppression of tumor growth in vivo by the mitocan alpha-tocopheryl succinate requires respiratory complex ii. Clin Cancer Res (2009) 15(5):1593–600. doi: 10.1158/1078-0432.CCR-08-2439
68. Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, et al. Repurposing atovaquone: Targeting mitochondrial complex iii and oxphos to eradicate cancer stem cells. Oncotarget (2016) 7(23):34084–99. doi: 10.18632/oncotarget.9122
69. Le SB, Hailer MK, Buhrow S, Wang Q, Flatten K, Pediaditakis P, et al. Inhibition of mitochondrial respiration as a source of adaphostin-induced reactive oxygen species and cytotoxicity. J Biol Chem (2007) 282(12):8860–72. doi: 10.1074/jbc.M611777200
70. Diepart C, Karroum O, Magat J, Feron O, Verrax J, Calderon PB, et al. Arsenic trioxide treatment decreases the oxygen consumption rate of tumor cells and radiosensitizes solid tumors. Cancer Res (2012) 72(2):482–90. doi: 10.1158/0008-5472.CAN-11-1755
71. Corso CR, Acco A, Bach C, Bonatto SJR, de Figueiredo BC, de Souza LM. Pharmacological profile and effects of mitotane in adrenocortical carcinoma. Br J Clin Pharmacol (2021) 87(7):2698–710. doi: 10.1111/bcp.14721
72. Rohlenova K, Sachaphibulkij K, Stursa J, Bezawork-Geleta A, Blecha J, Endaya B, et al. Selective disruption of respiratory supercomplexes as a new strategy to suppress Her2(High) breast cancer. Antioxid Redox Signal (2017) 26(2):84–103. doi: 10.1089/ars.2016.6677
73. Cheng G, Zielonka J, Ouari O, Lopez M, McAllister D, Boyle K, et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res (2016) 76(13):3904–15. doi: 10.1158/0008-5472.CAN-15-2534
74. Boukalova S, Stursa J, Werner L, Ezrova Z, Cerny J, Bezawork-Geleta A, et al. Mitochondrial targeting of metformin enhances its activity against pancreatic cancer. Mol Cancer Ther (2016) 15(12):2875–86. doi: 10.1158/1535-7163.MCT-15-1021
75. Dong LF, Jameson VJ, Tilly D, Cerny J, Mahdavian E, Marin-Hernandez A, et al. Mitochondrial targeting of vitamin e succinate enhances its pro-apoptotic and anti-cancer activity Via mitochondrial complex ii. J Biol Chem (2011) 286(5):3717–28. doi: 10.1074/jbc.M110.186643
76. Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: Zodiac-16. Diabetes Care (2010) 33(2):322–6. doi: 10.2337/dc09-1380
77. Pollak MN. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discovery (2012) 2(9):778–90. doi: 10.1158/2159-8290.CD-12-0263
78. McGuinness ME, Talbert RL. Phenformin-induced lactic acidosis: A forgotten adverse drug reaction. Ann Pharmacother (1993) 27(10):1183–7. doi: 10.1177/106002809302701004
79. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife (2014) 3:e02242. doi: 10.7554/eLife.02242
80. Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, et al. Dual inhibition of the lactate transporters Mct1 and Mct4 is synthetic lethal with metformin due to nad+ depletion in cancer cells. Cell Rep (2018) 25(11):3047–58 e4. doi: 10.1016/j.celrep.2018.11.043
81. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8(23):3984–4001. doi: 10.4161/cc.8.23.10238
82. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol (2017) 216(11):3799–816. doi: 10.1083/jcb.201704053
83. D'Antongiovanni V, Martinelli S, Richter S, Canu L, Guasti D, Mello T, et al. The microenvironment induces collective migration in sdhb-silenced mouse pheochromocytoma spheroids. Endocr Relat Cancer (2017) 24(10):555–64. doi: 10.1530/erc-17-0212
84. Martinelli S, Amore F, Mello T, Mannelli M, Maggi M, Rapizzi E. Metformin treatment induces different response in Pheochromocytoma/Paraganglioma tumour cells and in primary fibroblasts. Cancers (Basel) (2022) 14(14):3471. doi: 10.3390/cancers14143471
85. Martinelli S, Riverso M, Mello T, Amore F, Parri M, Simeone I, et al. Sdhb and sdhd silenced pheochromocytoma spheroids respond differently to tumour microenvironment and their aggressiveness is inhibited by impairing stroma metabolism. Mol Cell Endocrinol (2022) 547:111594. doi: 10.1016/j.mce.2022.111594
86. Shao S, Zhao L, An G, Zhang L, Jing X, Luo M, et al. Metformin suppresses hif-1alpha expression in cancer-associated fibroblasts to prevent tumor-stromal cross talk in breast cancer. FASEB J (2020) 34(8):10860–70. doi: 10.1096/fj.202000951RR
87. Xu S, Yang Z, Jin P, Yang X, Li X, Wei X, et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol Cancer Ther (2018) 17(6):1291–302. doi: 10.1158/1535-7163.MCT-17-0927
88. Chen G, Yu C, Tang Z, Liu S, An F, Zhu J, et al. Metformin suppresses gastric cancer progression through calmodulinlike protein 3 secreted from tumorassociated fibroblasts. Oncol Rep (2019) 41(1):405–14. doi: 10.3892/or.2018.6783
89. Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, Chandel NS. Mitochondrial complex iii is necessary for endothelial cell proliferation during angiogenesis. Nat Metab (2019) 1(1):158–71. doi: 10.1038/s42255-018-0011-x
90. Schiffmann LM, Werthenbach JP, Heintges-Kleinhofer F, Seeger JM, Fritsch M, Gunther SD, et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat Commun (2020) 11(1):3653. doi: 10.1038/s41467-020-17472-2
91. Magalhaes-Novais S, Blecha J, Naraine R, Mikesova J, Abaffy P, Pecinova A, et al. Mitochondrial respiration supports autophagy to provide stress resistance during quiescence. Autophagy (2022) 18(10):2409–26. doi: 10.1080/15548627.2022.2038898
92. Dong LF, Swettenham E, Eliasson J, Wang XF, Gold M, Medunic Y, et al. Vitamin e analogues inhibit angiogenesis by selective induction of apoptosis in proliferating endothelial cells: The role of oxidative stress. Cancer Res (2007) 67(24):11906–13. doi: 10.1158/0008-5472.CAN-07-3034
93. Rohlena J, Dong L-F, Kluckova K, Zobalova R, Goodwin J, Tilly D, et al. Mitochondrially targeted- A tocopheryl succinate is antiangiogenic: Potential benefit against tumor angiogenesis but caution against wound healing. Antioxidants Redox Signaling (2011) 15(12):2923–35. doi: 10.1089/ars.2011.4192
94. Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, et al. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell (2003) 3(5):497–509. doi: 10.1016/s1535-6108(03)00109-0
95. Han J, Li Y, Liu X, Zhou T, Sun H, Edwards P, et al. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PloS One (2018) 13(3):e0193031. doi: 10.1371/journal.pone.0193031
96. Dallaglio K, Bruno A, Cantelmo AR, Esposito AI, Ruggiero L, Orecchioni S, et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis (2014) 35(5):1055–66. doi: 10.1093/carcin/bgu001
97. Wang JC, Li GY, Wang B, Han SX, Sun X, Jiang YN, et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization Via pdgf-b downregulation. J Exp Clin Cancer Res (2019) 38(1):235. doi: 10.1186/s13046-019-1211-2
98. Li M, Yu X, Li W, Liu T, Deng G, Liu W, et al. Deguelin suppresses angiogenesis in human hepatocellular carcinoma by targeting hgf-C-Met pathway. Oncotarget (2018) 9(1):152–66. doi: 10.18632/oncotarget.22077
99. Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, et al. Mitochondrial complex iii is essential for suppressive function of regulatory T cells. Nature (2019) 565(7740):495–9. doi: 10.1038/s41586-018-0846-z
100. Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of Cd4(+)Cd25(+) regulatory T cells in the tumor microenvironment by metformin, a type 2 diabetes drug. EBioMedicine (2017) 25:154–64. doi: 10.1016/j.ebiom.2017.10.009
101. Stemberkova-Hubackova S, Zobalova R, Dubisova M, Smigova J, Dvorakova S, Korinkova K, et al. Simultaneous targeting of mitochondrial metabolism and immune checkpoints as a new strategy for renal cancer therapy. Clin Transl Med (2022) 12(3):e645. doi: 10.1002/ctm2.645
102. Liu Q, Tong D, Liu G, Gao J, Wang LA, Xu J, et al. Metformin inhibits prostate cancer progression by targeting tumor-associated inflammatory infiltration. Clin Cancer Res (2018) 24(22):5622–34. doi: 10.1158/1078-0432.CCR-18-0420
103. Wang JC, Sun X, Ma Q, Fu GF, Cong LL, Zhang H, et al. Metformin's antitumour and anti-angiogenic activities are mediated by skewing macrophage polarization. J Cell Mol Med (2018) 22(8):3825–36. doi: 10.1111/jcmm.13655
104. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U.S.A. (2015) 112(6):1809–14. doi: 10.1073/pnas.1417636112
105. Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity Via endoplasmic-Reticulum-Associated degradation of pd-L1. Mol Cell (2018) 71(4):606–20 e7. doi: 10.1016/j.molcel.2018.07.030
106. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in sdhd, a mitochondrial complex ii gene, in hereditary paraganglioma. Science (2000) 287(5454):848–51. doi: 10.1126/science.287.5454.848
107. Pasini B, Stratakis CA. Sdh mutations in tumorigenesis and inherited endocrine tumours: Lesson from the phaeochromocytoma-paraganglioma syndromes. J Internal Med (2009) 266(1):19–42. doi: 10.1111/j.1365-2796.2009.02111.x
108. Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci (2001) 98(6):3387–92. doi: 10.1073/pnas.051633798
109. Parsons DW, Jones SN, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321(5897):1807–12. doi: 10.1126/science.1164382
110. Hadrava Vanova K, Pang Y, Krobova L, Kraus M, Nahacka Z, Boukalova S, et al. Germline Suclg2 variants in patients with pheochromocytoma and paraganglioma. J Natl Cancer Inst (2022) 114(1):130–8. doi: 10.1093/jnci/djab158
111. Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of idh mutations in cancer. Cancer Cell (2018) 34(2):186–95. doi: 10.1016/j.ccell.2018.04.011
112. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep (2011) 12(5):463–9. doi: 10.1038/embor.2011.43
113. Stein EM, Fathi AT, DiNardo CD, Pollyea DA, Roboz GJ, Collins R, et al. Enasidenib in patients with mutant Idh2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, Ag221-C-001 trial. Lancet Haematol (2020) 7(4):e309–e19. doi: 10.1016/S2352-3026(19)30284-4
114. Stein EM, Dinardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant Idh2 relapsed or refractory acute myeloid leukemia. Blood (2017) 130(6):722–31. doi: 10.1182/blood-2017-04-779405
115. Mao MJ, Leonardi DE. Vascular-endothelial response to Idh1 mutant fibrosarcoma secretome and metabolite: Implications on cancer microenvironment. Am J Cancer Res (2019) 9(1):122–33.
116. Seok J, Yoon SH, Lee SH, Jung JH, Lee YM. The oncometabolite D−2−Hydroxyglutarate induces angiogenic activity through the vascular endothelial growth factor receptor 2 signaling pathway. Int J Oncol (2019) 54(2):753–63. doi: 10.3892/ijo.2018.4649
117. Zhang L, He L, Lugano R, Roodakker K, Bergqvist M, Smits A, et al. Idh mutation status is associated with distinct vascular gene expression signatures in lower-grade gliomas. Neuro-Oncology (2018) 20(11):1505–16. doi: 10.1093/neuonc/noy088
118. Xiang X, Liu Z, Zhang C, Li Z, Gao J, Zhang C, et al. Idh mutation subgroup status associates with intratumor heterogeneity and the tumor microenvironment in intrahepatic cholangiocarcinoma. Adv Sci (2021) 8(17):2101230. doi: 10.1002/advs.202101230
119. Chuntova P, Yamamichi A, Chen T, Narayanaswamy R, Ronseaux S, Hudson C, et al. Inhibition of d-2hg leads to upregulation of a proinflammatory gene signature in a novel hla-A2/Hla-Dr1 transgenic mouse model of Idh1r132h-expressing glioma. J ImmunoTher Cancer (2022) 10(5):e004644. doi: 10.1136/jitc-2022-004644
120. Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress Stat1 and Cd8+ T cell accumulation in gliomas. J Clin Invest (2017) 127(4):1425–37. doi: 10.1172/jci90644
121. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in idh-mutant gliomas by single-cell rna-seq. Science (2017) 355(6332):eaai8478. doi: 10.1126/science.aai8478
122. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
123. Yang C, Ko B, Hensley CT, Jiang L, Ajla T, Wasti, et al. Glutamine oxidation maintains the tca cycle and cell survival during impaired mitochondrial pyruvate transport. (2014) 56(3):414–24. doi: 10.1016/j.molcel.2014.09.025
124. Sappington DR, Siegel ER, Hiatt G, Desai A, Penney RB, Jamshidi-Parsian A, et al. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim Biophys Acta (BBA) - Gen Subj (2016) 1860(4):836–43. doi: 10.1016/j.bbagen.2016.01.021
125. Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther (1990) 46(2):243–71. doi: 10.1016/0163-7258(90)90094-i
126. Rais R, Jančařík A, Tenora L, Nedelcovych M, Alt J, Englert J, et al. Discovery of 6-Diazo-5-Oxo-L-Norleucine (Don) prodrugs with enhanced csf delivery in monkeys: A potential treatment for glioblastoma. J Med Chem (2016) 59(18):8621–33. doi: 10.1021/acs.jmedchem.6b01069
127. Oh M-H, Sun I-H, Zhao L, Leone RD, Sun I-M, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest (2020) 130(7):3865–84. doi: 10.1172/jci131859
128. Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (2019) 366(6468):1013–21. doi: 10.1126/science.aav2588
129. Yang L, Achreja A, Yeung T-L, Mangala Lingegowda S, Jiang D, Han C, et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab (2016) 24(5):685–700. doi: 10.1016/j.cmet.2016.10.011
130. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest (2021) 131(4):e140100. doi: 10.1172/jci140100
131. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of Asct2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med (2018) 24(2):194–202. doi: 10.1038/nm.4464
132. Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest (2015) 125(6):2293–306. doi: 10.1172/jci75836
133. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor cb-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.mct-13-0870
134. Robinson M,M, Mcbryant J,S, Tsukamoto T, Rojas C, Ferraris V,D, Hamilton K,S, et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-Phenylacetamido-1,2,4-Thiadiazol-2-Yl)Ethyl sulfide (Bptes). Biochem J (2007) 406(3):407–14. doi: 10.1042/bj20070039
135. Jin H, Wang S, Zaal EA, Wang C, Wu H, Bosma A, et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife (2020) 9:e56749. doi: 10.7554/elife.56749
136. Varghese S, Pramanik S, Williams LJ, Hodges HR, Hudgens CW, Fischer GM, et al. The glutaminase inhibitor cb-839 (Telaglenastat) enhances the antimelanoma activity of T-Cell–mediated immunotherapies. Mol Cancer Ther (2021) 20(3):500–11. doi: 10.1158/1535-7163.mct-20-0430
137. Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Negrón Abril YL, Goldford J, et al. Glutaminase inhibition impairs Cd8 T cell activation in Stk11-/Lkb1-Deficient lung cancer. Cell Metab (2022) 34(6):874–87.e6. doi: 10.1016/j.cmet.2022.04.003
138. Huang H, Vandekeere S, Kalucka J, Bierhansl L, Zecchin A, Brüning U, et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J (2017) 36(16):2334–52. doi: 10.15252/embj.201695518
139. Le A, Lane N,A, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism Via tca cycling for proliferation and survival in b cells. Cell Metab (2012) 15(1):110–21. doi: 10.1016/j.cmet.2011.12.009
140. Byun J-K, Park M, Lee S, Yun JW, Lee J, Kim JS, et al. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol Cell (2020) 80(4):592–606.e8. doi: 10.1016/j.molcel.2020.10.015
141. Elgogary A, Xu Q, Poore B, Alt J, Zimmermann SC, Zhao L, et al. Combination therapy with bptes nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci (2016) 113(36):E5328–E36. doi: 10.1073/pnas.1611406113
142. Eelen G, Dubois C, Cantelmo AR, Goveia J, Brüning U, Deran M, et al. Role of glutamine synthetase in angiogenesis beyond glutamine synthesis. Nature (2018) 561(7721):63–9. doi: 10.1038/s41586-018-0466-7
143. Palmieri EM, Menga A, Martín-Pérez R, Quinto A, Riera-Domingo C, De Tullio G, et al. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep (2017) 20(7):1654–66. doi: 10.1016/j.celrep.2017.07.054
144. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discovery (2022) 21(2):141–62. doi: 10.1038/s41573-021-00339-6
145. Kidd JG. Regression of transplanted lymphomas induced in vivo by means of normal Guinea pig serum. J Exp Med (1953) 98(6):583–606. doi: 10.1084/jem.98.6.583
146. Broome JD. Evidence that the l-asparaginase of Guinea pig serum is responsible for its antilymphoma effects. J Exp Med (1963) 118(1):121–48. doi: 10.1084/jem.118.1.121
147. Chiu M, Taurino G, Bianchi MG, Kilberg MS, Bussolati O. Asparagine synthetase in cancer: Beyond acute lymphoblastic leukemia. Front Oncol (2019) 9:1480. doi: 10.3389/fonc.2019.01480
148. Maxwell M, McCoy TA, Neuman RE. The amino acid requirements of the walker carcinosarcoma 256 in vitro. Cancer Res (1956) 16(10 Part 1):979–84.
149. Wu J, Li G, Li L, Li D, Dong Z, Jiang P. Asparagine enhances lck signalling to potentiate Cd8+ T-cell activation and anti-tumour responses. Nat Cell Biol (2021) 23(1):75–86. doi: 10.1038/s41556-020-00615-4
150. Alexandrou C, Al-Aqbi SS, Higgins JA, Boyle W, Karmokar A, Andreadi C, et al. Sensitivity of colorectal cancer to arginine deprivation therapy is shaped by differential expression of urea cycle enzymes. Sci Rep (2018) 8(1):12096. doi: 10.1038/s41598-018-30591-7
151. Albaugh VL, Pinzon-Guzman C, Barbul A. Arginine-dual roles as an onconutrient and immunonutrient. J Surg Oncol (2017) 115(3):273–80. doi: 10.1002/jso.24490
152. Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood (2015) 125(15):2386–96. doi: 10.1182/blood-2014-09-600643
153. Fultang L, Booth S, Yogev O, Martins Da Costa B, Tubb V, Panetti S, et al. Metabolic engineering against the arginine microenvironment enhances car-T cell proliferation and therapeutic activity. Blood (2020) 136(10):1155–60. doi: 10.1182/blood.2019004500
154. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
155. Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature (2021) 598(7882):662–6. doi: 10.1038/s41586-021-04003-2
156. Kaiser P. Methionine dependence of cancer. Biomolecules (2020) 10(4):568. doi: 10.3390/biom10040568
157. Sanderson SM, Gao X, Dai Z, Locasale JW. Methionine metabolism in health and cancer: A nexus of diet and precision medicine. Nat Rev Cancer (2019) 19(11):625–37. doi: 10.1038/s41568-019-0187-8
158. Hoffman RM, Tan Y, Li S, Han Q, Zavala J, Zavala J Jr. Pilot phase I clinical trial of methioninase on high-stage cancer patients: Rapid depletion of circulating methionine. Methods Mol Biol (2019) 1866:231–42 doi: 10.1007/978-1-4939-8796-2_17
159. Hung MH, Lee JS, Ma C, Diggs LP, Heinrich S, Chang CW, et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun (2021) 12(1):1455. doi: 10.1038/s41467-021-21804-1
160. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer Slc43a2 alters T cell methionine metabolism and histone methylation. Nature (2020) 585(7824):277–82. doi: 10.1038/s41586-020-2682-1
161. Burke L, Guterman I, Palacios Gallego R, Britton RG, Burschowsky D, Tufarelli C, et al. The janus-like role of proline metabolism in cancer. Cell Death Discovery (2020) 6:104. doi: 10.1038/s41420-020-00341-8
162. Loayza-Puch F, Rooijers K, Buil LCM, Zijlstra J, F. Oude Vrielink J, Lopes R, et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature (2016) 530(7591):490–4. doi: 10.1038/nature16982
163. Westbrook RL, Bridges E, Roberts J, Escribano-Gonzalez C, Eales KL, Vettore LA, et al. Proline synthesis through Pycr1 is required to support cancer cell proliferation and survival in oxygen-limiting conditions. Cell Rep (2022) 38(5):110320. doi: 10.1016/j.celrep.2022.110320
164. Liu Y, Mao C, Wang M, Liu N, Ouyang L, Liu S, et al. Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene (2020) 39(11):2358–76. doi: 10.1038/s41388-019-1151-5
165. Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun (2017) 8(1):15267. doi: 10.1038/ncomms15267
166. Bai J, Liu T, Tu B, Yuan M, Shu Z, Fan M, et al. Autophagy loss impedes cancer-associated fibroblast activation via downregulating proline biosynthesis. Autophagy (2022) 1-12. doi: 10.1080/15548627.2022.2093026
167. Milne K, Sun J, Zaal EA, Mowat J, Celie PHN, Fish A, et al. A fragment-like approach to Pycr1 inhibition. Bioorg Med Chem Lett (2019) 29(18):2626–31. doi: 10.1016/j.bmcl.2019.07.047
168. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis (2016) 5:e189. doi: 10.1038/oncsis.2015.49
169. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1c promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev (2011) 25(10):1041–51. doi: 10.1101/gad.1987211
170. Schcolnik-Cabrera A, Chavez-Blanco A, Dominguez-Gomez G, Taja-Chayeb L, Morales-Barcenas R, Trejo-Becerril C, et al. Orlistat as a fasn inhibitor and multitargeted agent for cancer therapy. Expert Opin Investig Drugs (2018) 27(5):475–89. doi: 10.1080/13543784.2018.1471132
171. Murata S, Yanagisawa K, Fukunaga K, Oda T, Kobayashi A, Sasaki R, et al. Fatty acid synthase inhibitor cerulenin suppresses liver metastasis of colon cancer in mice. Cancer Sci (2010) 101(8):1861–5. doi: 10.1111/j.1349-7006.2010.01596.x
172. Alli PM, Pinn ML, Jaffee EM, McFadden JM, Kuhajda FP. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-n transgenic mice. Oncogene (2005) 24(1):39–46. doi: 10.1038/sj.onc.1208174
173. Li S, Qiu L, Wu B, Shen H, Zhu J, Zhou L, et al. Tofa suppresses ovarian cancer cell growth in vitro and in vivo. Mol Med Rep (2013) 8(2):373–8. doi: 10.3892/mmr.2013.1505
174. Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid catabolism Via Cpt1 as a therapeutic target for prostate cancer. Mol Cancer Ther (2014) 13(10):2361–71. doi: 10.1158/1535-7163.MCT-14-0183
175. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest (2010) 120(1):142–56. doi: 10.1172/JCI38942
176. Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P, et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ergo (Etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond) (2007) 113(4):205–12. doi: 10.1042/CS20060307
177. Pacilli A, Calienni M, Margarucci S, D'Apolito M, Petillo O, Rocchi L, et al. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J Natl Cancer Inst (2013) 105(7):489–98. doi: 10.1093/jnci/djt030
178. McKelvey KJ, Wilson EB, Short S, Melcher AA, Biggs M, Diakos CI, et al. Glycolysis and fatty acid oxidation inhibition improves survival in glioblastoma. Front Oncol (2021) 11:633210. doi: 10.3389/fonc.2021.633210
179. Lopes-Coelho F, Andre S, Felix A, Serpa J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol Cell Endocrinol (2018) 462(Pt B):93–106. doi: 10.1016/j.mce.2017.01.031
180. Santi A, Caselli A, Ranaldi F, Paoli P, Mugnaioni C, Michelucci E, et al. Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim Biophys Acta (2015) 1853(12):3211–23. doi: 10.1016/j.bbamcr.2015.09.013
181. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X, et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis (2020) 11(4):267. doi: 10.1038/s41419-020-2434-z
182. Peng S, Chen D, Cai J, Yuan Z, Huang B, Li Y, et al. Enhancing cancer-associated fibroblast fatty acid catabolism within a metabolically challenging tumor microenvironment drives colon cancer peritoneal metastasis. Mol Oncol (2021) 15(5):1391–411. doi: 10.1002/1878-0261.12917
183. Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty acid carbon is essential for dntp synthesis in endothelial cells. Nature (2015) 520(7546):192–7. doi: 10.1038/nature14362
184. Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature (2017) 542(7639):49–54. doi: 10.1038/nature21028
185. Patella F, Schug ZT, Persi E, Neilson LJ, Erami Z, Avanzato D, et al. Proteomics-based metabolic modeling reveals that fatty acid oxidation (Fao) controls endothelial cell (Ec) permeability. Mol Cell Proteomics (2015) 14(3):621–34. doi: 10.1074/mcp.M114.045575
186. Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Bruning U, et al. Quiescent endothelial cells upregulate fatty acid beta-oxidation for vasculoprotection Via redox homeostasis. Cell Metab (2018) 28(6):881–94 e13. doi: 10.1016/j.cmet.2018.07.016
187. Wei X, Schneider JG, Shenouda SM, Lee A, Towler DA, Chakravarthy MV, et al. De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (Enos) palmitoylation. J Biol Chem (2011) 286(4):2933–45. doi: 10.1074/jbc.M110.193037
188. Seguin F, Carvalho MA, Bastos DC, Agostini M, Zecchin KG, Alvarez-Flores MP, et al. The fatty acid synthase inhibitor orlistat reduces experimental metastases and angiogenesis in B16-F10 melanomas. Br J Cancer (2012) 107(6):977–87. doi: 10.1038/bjc.2012.355
189. Browne CD, Hindmarsh EJ, Smith JW. Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. FASEB J (2006) 20(12):2027–35. doi: 10.1096/fj.05-5404com
190. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab (2012) 15(4):432–7. doi: 10.1016/j.cmet.2011.11.013
191. Namgaladze D, Lips S, Leiker TJ, Murphy RC, Ekroos K, Ferreiros N, et al. Inhibition of macrophage fatty acid beta-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia (2014) 57(5):1067–77. doi: 10.1007/s00125-014-3173-4
192. Xiu F, Diao L, Qi P, Catapano M, Jeschke MG. Palmitate differentially regulates the polarization of differentiating and differentiated macrophages. Immunology (2016) 147(1):82–96. doi: 10.1111/imm.12543
193. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med (2019) 11(11):e10698. doi: 10.15252/emmm.201910698
194. Bose D, Banerjee S, Chatterjee N, Das S, Saha M, Saha KD. Inhibition of tgf-beta induced lipid droplets switches M2 macrophages to M1 phenotype. Toxicol In Vitro (2019) 58:207–14. doi: 10.1016/j.tiv.2019.03.037
195. Schumann T, Adhikary T, Wortmann A, Finkernagel F, Lieber S, Schnitzer E, et al. Deregulation of Pparbeta/Delta target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget (2015) 6(15):13416–33. doi: 10.18632/oncotarget.3826
196. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog Lipid Res (2020) 80:101055. doi: 10.1016/j.plipres.2020.101055
197. Kurniawan H, Soriano-Baguet L, Brenner D. Regulatory T cell metabolism at the intersection between autoimmune diseases and cancer. Eur J Immunol (2020) 50(11):1626–42. doi: 10.1002/eji.201948470
198. Kleinfeld AM, Okada C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-Lymphocyte-Mediated killing. J Lipid Res (2005) 46(9):1983–90. doi: 10.1194/jlr.M500151-JLR200
199. Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, et al. Hif-1alpha is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep (2019) 27(1):226–37 e4. doi: 10.1016/j.celrep.2019.03.029
200. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. Stat3 activation-induced fatty acid oxidation in Cd8(+) T effector cells is critical for obesity-promoted breast tumor growth. Cell Metab (2020) 31(1):148–61 e5. doi: 10.1016/j.cmet.2019.10.013
201. Chowdhury PS, Chamoto K, Kumar A, Honjo T. Ppar-induced fatty acid oxidation in T cells increases the number of tumor-reactive Cd8(+) T cells and facilitates anti-Pd-1 therapy. Cancer Immunol Res (2018) 6(11):1375–87. doi: 10.1158/2326-6066.CIR-18-0095
202. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
203. de Almeida LY, Mariano FS, Bastos DC, Cavassani KA, Raphelson J, Mariano VS, et al. The antimetastatic activity of orlistat is accompanied by an antitumoral immune response in mouse melanoma. Cancer Chemother Pharmacol (2020) 85(2):321–30. doi: 10.1007/s00280-019-04010-1
204. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/sciadv.1600200
205. Wang W, Cui J, Ma H, Lu W, Huang J. Targeting pyrimidine metabolism in the era of precision cancer medicine. Front Oncol (2021) 11:684961. doi: 10.3389/fonc.2021.684961
206. Ogrodzinski MP, Teoh ST, Lunt SY. Targeting subtype-specific metabolic preferences in nucleotide biosynthesis inhibits tumor growth in a breast cancer model. Cancer Res (2021) 81(2):303–14. doi: 10.1158/0008-5472.can-20-1666
207. Farber S, Diamond LK, Mercer RD, Sylvester RF, Wolff JA. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-Aminopteroyl-Glutamic acid (Aminopterin). New Engl J Med (1948) 238(23):787–93. doi: 10.1056/nejm194806032382301
208. Walling J. From methotrexate to pemetrexed and beyond. a review of the pharmacodynamic and clinical properties of antifolates. Investigat New Drugs (2006) 24(1):37–77. doi: 10.1007/s10637-005-4541-1
209. Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol (2017) 24(9):1161–80. doi: 10.1016/j.chembiol.2017.08.028
210. An Q, Robins P, Lindahl T, Barnes DE. 5-fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res (2007) 67(3):940–5. doi: 10.1158/0008-5472.can-06-2960
211. Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin Oncol (1995) 22(4 Suppl 11):3–10.
212. Boukalova S, Hubackova S, Milosevic M, Ezrova Z, Neuzil J, Rohlena J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim Biophys Acta Mol Basis Dis (2020) 1866(6):165759. doi: 10.1016/j.bbadis.2020.165759
213. Zhou Y, Tao L, Zhou X, Zuo Z, Gong J, Liu X, et al. Dhodh and cancer: Promising prospects to be explored. Cancer Metab (2021) 9(1):22. doi: 10.1186/s40170-021-00250-z
214. Li L, Ng SR, Colon CI, Drapkin BJ, Hsu PP, Li Z, et al. Identification of dhodh as a therapeutic target in small cell lung cancer. Sci Transl Med (2019) 11(517):eaaw7852. doi: 10.1126/scitranslmed.aaw7852
215. Mathur D, Stratikopoulos E, Ozturk S, Steinbach N, Pegno S, Schoenfeld S, et al. Pten regulates glutamine flux to pyrimidine synthesis and sensitivity to dihydroorotate dehydrogenase inhibition. Cancer Discovery (2017) 7(4):380–90. doi: 10.1158/2159-8290.CD-16-0612
216. Koundinya M, Sudhalter J, Courjaud A, Lionne B, Touyer G, Bonnet L, et al. Dependence on the pyrimidine biosynthetic enzyme dhodh is a synthetic lethal vulnerability in mutant kras-driven cancers. Cell Chem Biol (2018) 25(6):705–17.e11. doi: 10.1016/j.chembiol.2018.03.005
217. Walter M, Herr P. Re-discovery of pyrimidine salvage as target in cancer therapy. Cells (2022) 11(4):739. doi: 10.3390/cells11040739
218. Cuthbertson CR, Guo H, Kyani A, Madak JT, Arabzada Z, Neamati N. The dihydroorotate dehydrogenase inhibitor brequinar is synergistic with Ent1/2 inhibitors. ACS Pharmacol Trans Sci (2020) 3(6):1242–52. doi: 10.1021/acsptsci.0c00124
219. Peters GJ, Schwartsmann G, Nadal JC, Laurensse EJ, van Groeningen CJ, van der Vijgh WJ, et al. In vivo inhibition of the pyrimidine De novo enzyme dihydroorotic acid dehydrogenase by brequinar sodium (Dup-785; nsc 368390) in mice and patients. Cancer Res (1990) 50(15):4644–9.
220. Dey P, Kimmelman AC, DePinho RA. Metabolic codependencies in the tumor microenvironment. Cancer Discovery (2021) 11(5):1067–81. doi: 10.1158/2159-8290.CD-20-1211
221. Rohlenova K, Goveia J, Garcia-Caballero M, Subramanian A, Kalucka J, Treps L, et al. Single-cell rna sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab (2020) 31(4):862–77 e14. doi: 10.1016/j.cmet.2020.03.009
222. Basaki Y, Chikahisa L, Aoyagi K, Miyadera K, Yonekura K, Hashimoto A, et al. Gamma-hydroxybutyric acid and 5-fluorouracil, metabolites of uft, inhibit the angiogenesis induced by vascular endothelial growth factor. Angiogenesis (2001) 4(3):163–73. doi: 10.1023/a:1014059528046
223. Yonekura K, Basaki Y, Chikahisa L, Okabe S, Hashimoto A, Miyadera K, et al. Uft and its metabolites inhibit the angiogenesis induced by murine renal cell carcinoma, as determined by a dorsal air sac assay in mice. Clin Cancer Res (1999) 5(8):2185–91.
224. Presta M, Belleri M, Vacca A, Ribatti D. Anti-angiogenic activity of the purine analog 6-thioguanine. Leukemia (2002) 16(8):1490–9. doi: 10.1038/sj.leu.2402646
225. Wang C, Xi W, Jiang J, Ji J, Yu Y, Zhu Z, et al. Metronomic chemotherapy remodel cancer-associated fibroblasts to decrease chemoresistance of gastric cancer in nude mice. Oncol Lett (2017) 14(6):7903–9. doi: 10.3892/ol.2017.7197
226. Lotti F, Jarrar AM, Pai RK, Hitomi M, Lathia J, Mace A, et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by il-17a. J Exp Med (2013) 210(13):2851–72. doi: 10.1084/jem.20131195
227. Quéméneur L, Gerland L-M, Flacher M, Ffrench M, Revillard J-P, Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J Immunol (2003) 170(10):4986–95. doi: 10.4049/jimmunol.170.10.4986
228. Peeters MJW, Aehnlich P, Pizzella A, Mølgaard K, Seremet T, Met Ö, et al. Mitochondrial-linked De novo pyrimidine biosynthesis dictates human T-cell proliferation but not expression of effector molecules. Front Immunol (2021) 12:718863. doi: 10.3389/fimmu.2021.718863
229. Siemasko KF, Chong ASF, Williams JW, Bremer EG, Finnegan A. Regulation of b cell function by the immunosuppressive agent Leflunomide1. Transplantation (1996) 61(4):635–42. doi: 10.1097/00007890-199602270-00020
230. Ringshausen I, Oelsner M, Bogner C, Peschel C, Decker T. The immunomodulatory drug leflunomide inhibits cell cycle progression of b-cll cells. Leukemia (2008) 22(3):635–8. doi: 10.1038/sj.leu.2404922
231. Chong AS, Finnegan A, Jiang X, Gebel H, Sankary HN, Foster P, et al. Leflunomide, a novel immunosuppressive agent. the mechanism of inhibition of T cell proliferation. Transplantation (1993) 55(6):1361–6. doi: 10.1097/00007890-199306000-00028
232. Read SW, DeGrezia M, Ciccone EJ, DerSimonian R, Higgins J, Adelsberger JW, et al. The effect of leflunomide on cycling and activation of T-cells in hiv-1-Infected participants. PloS One (2010) 5(8):e11937. doi: 10.1371/journal.pone.0011937
233. Pinschewer DD, Ochsenbein AF, Fehr T, Zinkernagel RM. Leflunomide-mediated suppression of antiviral antibody and T cell responses: Differential restoration by Uridine1. Transplantation (2001) 72(4):712–9. doi: 10.1097/00007890-200108270-00026
234. Aly L, Hemmer B, Korn T. From leflunomide to teriflunomide: Drug development and immunosuppressive oral drugs in the treatment of multiple sclerosis. Curr Neuropharmacol (2017) 15(6):874–91. doi: 10.2174/1570159X14666161208151525
235. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70(8):3052–61. doi: 10.1158/0008-5472.CAN-09-3690
236. Tian J, Zhang D, Kurbatov V, Wang Q, Wang Y, Fang D, et al. 5-fluorouracil efficacy requires anti-tumor immunity triggered by cancer-Cell-Intrinsic sting. EMBO J (2021) 40(7):e106065. doi: 10.15252/embj.2020106065
237. Wu Y, Deng Z, Wang H, Ma W, Zhou C, Zhang S. Repeated cycles of 5-fluorouracil chemotherapy impaired anti-tumor functions of cytotoxic T cells in a Ct26 tumor-bearing mouse model. BMC Immunol (2016) 17(1):29. doi: 10.1186/s12865-016-0167-7
238. Yusung S, McGovern D, Lin L, Hommes D, Lagishetty V, Braun J. Nk cells are biologic and biochemical targets of 6-mercaptopurine in crohn's disease patients. Clin Immunol (2017) 175:82–90. doi: 10.1016/j.clim.2016.12.004
239. McCarthy MT, Moncayo G, Hiron TK, Jakobsen NA, Valli A, Soga T, et al. Purine nucleotide metabolism regulates expression of the human immune ligand mica. J Biol Chem (2018) 293(11):3913–24. doi: 10.1074/jbc.m117.809459
240. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi: 10.1038/nri2216
241. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic gr-1+/Cd11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11(18):6713–21. doi: 10.1158/1078-0432.ccr-05-0883
242. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in Balb/C mice bearing 4t1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol (2009) 9(7-8):900–9. doi: 10.1016/j.intimp.2009.03.015
243. Coelho AR, Oliveira PJ. Dihydroorotate dehydrogenase inhibitors in sars-Cov-2 infection. Eur J Clin Invest (2020) 50(10):e13366. doi: 10.1111/eci.13366
244. Sprenger H-G, Macvicar T, Bahat A, Fiedler KU, Hermans S, Ehrentraut D, et al. Cellular pyrimidine imbalance triggers mitochondrial DNA–dependent innate immunity. Nat Metab (2021) 3(5):636–50. doi: 10.1038/s42255-021-00385-9
245. Lu C-S, Lin C-W, Chang Y-H, Chen H-Y, Chung W-C, Lai W-Y, et al. Antimetabolite pemetrexed primes a favorable tumor microenvironment for immune checkpoint blockade therapy. J ImmunoTher Cancer (2020) 8(2):e001392. doi: 10.1136/jitc-2020-001392
Keywords: cancer, metabolism, tumor micro environment (TME), glycolysis, oxidative phoshorylation, fatty acid metabolism, nucleotide metabolism, endothelial cells
Citation: Hyroššová P, Milošević M, Škoda J, Vachtenheim Jr J, Rohlena J and Rohlenová K (2022) Effects of metabolic cancer therapy on tumor microenvironment. Front. Oncol. 12:1046630. doi: 10.3389/fonc.2022.1046630
Received: 16 September 2022; Accepted: 28 November 2022;
Published: 13 December 2022.
Edited by:
Sara Rodriguez-Enriquez, Facultad de Estudios Superiores Iztacala, MexicoCopyright © 2022 Hyroššová, Milošević, Škoda, Vachtenheim Jr, Rohlena and Rohlenová. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kateřina Rohlenová, katerina.rohlenova@ibt.cas.cz; Jakub Rohlena, jakub.rohlena@ibt.cas.cz