


Positive Effect of Acetylation on Proteomic Analysis Based on Liquid Chromatography with Atmospheric Pressure Chemical Ionization and Photoionization Mass Spectrometry

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Mass Spectra of Acetylated Peptide Standards
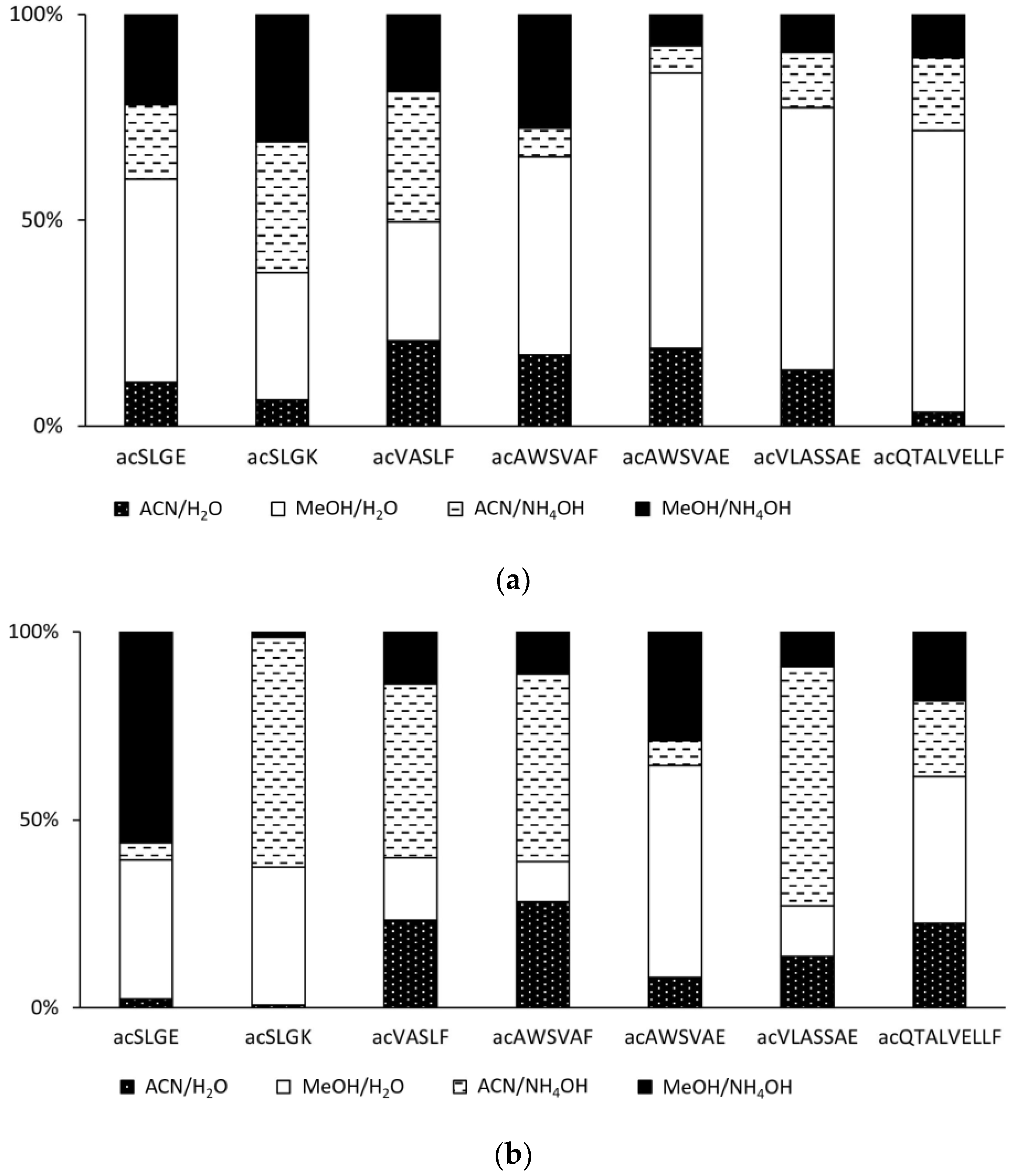
2.2. Effect of the Mobile Phase on the Ionization of Acetylated Peptides
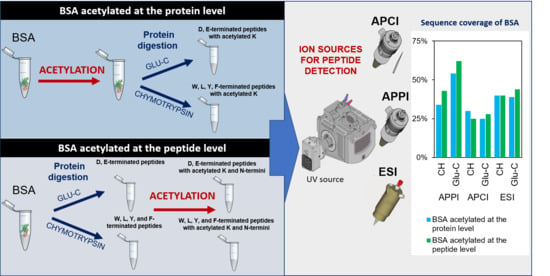
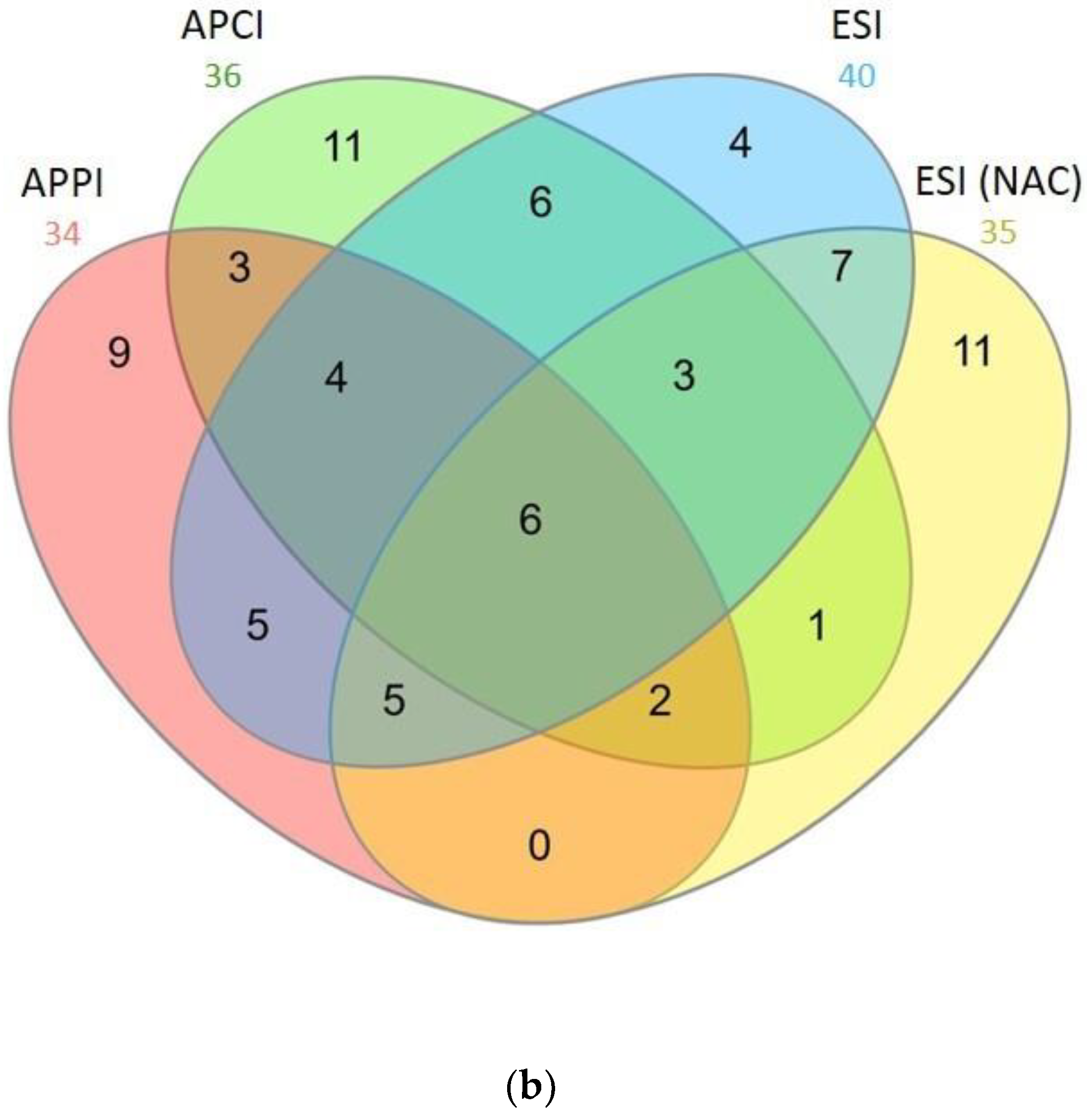
2.3. Bottom-Up Proteomics of BSA Digests
3. Materials and Methods
3.1. Chemicals and Reagent
3.2. Peptide Standards
3.3. Protein Digestion
3.4. Acetylation
3.5. Mass Spectrometry
3.6. Flow Injection Analysis
3.7. Liquid Chromatography-Tandem Mass Spectrometry
3.8. Data Interpretation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R. Direct Analysis of Protein Complexes Using Mass Spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative Analysis of Complex Protein Mixtures Using Isotope-Coded Affinity Tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef]
- Vit, O.; Petrak, J. Integral Membrane Proteins in Proteomics. How to Break Open the Black Box? J. Proteom. 2017, 153, 8–20. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Feist, P.; Hummon, A.B. Proteomic Challenges: Sample Preparation Techniques for Microgram-Quantity Protein Analysis from Biological Samples. Int. J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef]
- Fischer, F.; Poetsch, A. Protein Cleavage Strategies for an Improved Analysis of the Membrane Proteome. Proteome Sci. 2006, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Chen, D.A.; Regnier, F.E. Rapid Verification of Disulfide Linkages in Recombinant Human Growth Hormone by Tandem Column Tryptic Mapping. J. Chromatogr. A 1998, 808, 121–131. [Google Scholar] [CrossRef]
- Julka, S.; Regnier, F.E. Benzoyl Derivatization as a Method to Improve Retention of Hydrophilic Peptides in Tryptic Peptide Mapping. Anal. Chem. 2004, 76, 5799–5806. [Google Scholar] [CrossRef] [PubMed]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An Overview of Matrix Effects in Liquid Chrmatography-Mass Spectrometry. Mass Spectrom. 2010, 30, 491–509. [Google Scholar] [CrossRef]
- Loo, R.R.O.; Dales, N.; Andrews, P.C. Surfactant Effects on Protein Structure Examined by Electrospray Ionization Mass Spectrometry. Protein Sci. 1994, 3, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Conrads, T.P.; Yu, L.; Terunuma, A.; Janini, G.M.; Issaq, H.J.; Vogel, J.C.; Veenstra, T.D. A Detergent- and Cyanogen Bromide-Free Method for Integral Membrane Proteomics: Application to Halobacterium Purple Membranes and The Human Epidermal Membrane Proteome. Proteomics 2004, 4, 31–45. [Google Scholar] [CrossRef]
- Xu, T.; Wang, H.; Wu, M.; Wang, W.; Tan, Q.; Zhao, F.; Zhou, F.; Hu, T.; Jiang, Z.; Liu, Z.; et al. Disulfide-Containing Detergents (DCDs) for the Structural Biology of Membrane Proteins Dongxiang. Chemistry 2019, 25, 11635–11640. [Google Scholar] [CrossRef]
- Donoghue, P.M.; Hughes, C.; Vissers, J.P.C.; Langridge, J.I.; Dunn, M.J. Nonionic Detergent Phase Extraction for the Proteomic Analysis of Heart Membrane Proteins Using Label-Free LC-MS. Proteomics 2008, 8, 3895–3905. [Google Scholar] [CrossRef] [PubMed]
- Breyton, C.; Pucci, B.; Popot, J. Amphiols and Fluorinated Surfactants: Two Alternatives to Detergents for Studying Membrane Proteins In Vitro. In Heterologous Expression of Membrane Proteins: Methods in Molecular Biology; Mus-Vetau, I., Ed.; Humana Press: Totowa, NJ, USA, 2010; Volume 601, pp. 219–245. ISBN 9781607613442. [Google Scholar]
- Wehbie, M.; Onyia, K.K.; Mahler, F.; Le Roy, A.; Deletraz, A.; Bouchemal, I.; Vargas, C.; Babalola, J.O.; Breyton, C.; Ebel, C.; et al. Maltose-Based Fluorinated Surfactants for Membrane-Protein Extraction and Stabilization. Langmuir 2021, 37, 2111–2122. [Google Scholar] [CrossRef]
- Gilmore, J.M.; Kettenbach, A.N.; Gerber, S.A. Increasing Phosphoproteomic Coverage through Sequential Digestion by Complementary Proteases. Anal. Bioanal. Chem. 2012, 402, 711–720. [Google Scholar] [CrossRef]
- Rietschel, B.; Bornemann, S.; Arrey, T.N.; Baeumlisberger, D.; Karas, M.; Meyer, B. Membrane Protein Analysis Using an Improved Peptic In-Solution Digestion Protocol. Proteomics 2009, 9, 5553–5557. [Google Scholar] [CrossRef]
- Wu, C.C.; MacCoss, M.J.; Howell, K.E.; Yates, J.R. A Method for the Comprehensive Proteomic Analysis of Membrane Proteins. Nat. Biotechnol. 2003, 21, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, Z.; Cupp-Sutton, K.A.; Liu, X.; Wu, S. Deep Intact Proteoform Characterization in Human Cell Lysate Using High-PH and Low-PH Reversed-Phase Liquid Chromatography. J. Am. Soc. Mass Spectrom. 2019, 30, 2502–2513. [Google Scholar] [CrossRef]
- Petritis, K.; Brussaux, S.; Guenu, S.; Elfakir, C.; Dreux, M. Ion-Pair Reversed-Phase Liquid Chromatography-Electrospray Mass Spectrometry for the Analysis of Underivatized Small Peptides. J. Chromatogr. A 2002, 957, 173–185. [Google Scholar] [CrossRef]
- Kalghatgi, K.; Horvath, C. Rapid Peptide Mapping by High-Performance Liquid Chromatography. J. Chromatogr. 1988, 443, 343–354. [Google Scholar] [CrossRef]
- Gustavsson, S.Å.; Samskog, J.; Markides, K.E.; Långström, B. Studies of Signal Suppression in Liquid Chromatography-Electrospray Ionization Mass Spectrometry Using Volatile Ion-Pairing Reagents. J. Chromatogr. A 2001, 937, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bonmatin, J.M.; Moineau, I.; Charvet, R.; Fleche, C.; Colin, M.E.; Bengsch, E.R. A LC/APCI-MS/MS Method for Analysis of Imidacloprid in Soils, in Plants, and in Pollens. Anal. Chem. 2003, 75, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E. The Potential of Organic (Electrospray- and Atmospheric Pressure Chemical Ionisation) Mass Spectrometric Techniques Coupled to Liquid-Phase Separation for Speciation Analysis. J. Chromatogr. A 2003, 1000, 841–889. [Google Scholar] [CrossRef]
- Raffaelli, A.; Saba, A. Atmospheric Pressure Photoionization Mass Spectrometry. Mass Spectrom. Rev. 2003, 22, 318–331. [Google Scholar] [CrossRef]
- Cristoni, S.; Bernardi, L.R.; Biunno, I.; Guidugli, F. Analysis of Peptides Using Partial (No Discharge) Atmospheric Pressure Chemical Ionization Conditions with Ion Trap Mass Spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Robb, D.B.; Blades, M.W. State-of-the-Art in Atmospheric Pressure Photoionization for LC/MS. Anal. Chim. Acta 2008, 627, 34–49. [Google Scholar] [CrossRef]
- Delobel, A.; Halgand, F.; Laffranchise-Gosse, B.; Snijders, H.; Laprévote, O. Characterization of Hydrophobic Peptides by Atmospheric Pressure Photoionization-Mass Spectrometry and Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 5961–5968. [Google Scholar] [CrossRef] [PubMed]
- Debois, D.; Giuliani, A.; Laprévote, O. Fragmentation Induced in Atmospheric Pressure Photoionization of Peptides. J. Mass Spectrom. 2006, 41, 1554–1560. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Matrix Effect in Quantitative LC/MS/MS Analyses of Biological Fluids: A Method for Determination of Finasteride in Human Plasma at Picogram Per Milliliter Concentrations. Anal. Chem. 1998, 70, 882–889. [Google Scholar] [CrossRef]
- Hsieh, Y.; Chintala, M.; Mei, H.; Agans, J.; Brisson, J.M.; Ng, K.; Korfmacher, W.A. Quantitative Screening and Matrix Effect Studies of Drug Discovery Compounds in Monkey Plasma Using Fastgradient Liquid Chromatography/Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2001, 15, 2481–2487. [Google Scholar] [CrossRef]
- McCulloch, R.D.; Robb, D.B. Field-Free Atmospheric Pressure Photoionization-Liquid Chromatography-Mass Spectrometry for the Analysis of Steroids within Complex Biological Matrices. Anal. Chem. 2017, 89, 4169–4176. [Google Scholar] [CrossRef]
- Shen, Y.; Han, C.; Chen, J.; Wang, X. Analysis of Cyclic Peptides in Pseudostellaria Heterophylla (Miq.) Pax by HPLC-APCI-MS. Chromatographia 2007, 66, 319–323. [Google Scholar] [CrossRef]
- Bose, U.; Hodson, M.; Shaw, P.; Fuerst, J.; Hewavitharana, A. Two Peptides, Cycloaspeptide A and Nazumamide A from a Sponge Associated Marine Actinobacterium Salinispora Sp. Nat. Prod. Commun. 2014, 9, 545–546. [Google Scholar] [CrossRef]
- Bagag, A.; Jault, J.M.; Sidahmed-Adrar, N.; Réfrégiers, M.; Giuliani, A.; Le Naour, F. Characterization of Hydrophobic Peptides in the Presence of Detergent by Photoionization Mass Spectrometry. PLoS ONE 2013, 8, e79033. [Google Scholar] [CrossRef] [PubMed]
- Bagag, A.; Giuliani, A.; Laprévote, O. Atmospheric Pressure Photoionization of Peptides. Int. J. Mass Spectrom. 2011, 299, 1–4. [Google Scholar] [CrossRef]
- Bagag, A.; Giuliani, A.; Réfrégiers, M.; Le Naour, F. Atmospheric Pressure Photoionization Study of Post-Translational Modifications: The Case of Palmitoylation. Int. J. Mass Spectrom. 2012, 328–329, 23–27. [Google Scholar] [CrossRef]
- Giuliani, A.; Giorgetta, J.L.; Ricaud, J.P.; Jamme, F.; Rouam, V.; Wien, F.; Laprévote, O.; Réfrégiers, M. Atmospheric Pressure Photoionization Using Tunable VUV Synchrotron Radiation. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2012, 279, 114–117. [Google Scholar] [CrossRef]
- Sedláčková, S.; Hubálek, M.; Vrkoslav, V.; Blechová, M.; Cvačka, J. Utility of Atmospheric-Pressure Chemical Ionization and Photoionization Mass Spectrometry in Bottom-Up Proteomics. Separations 2022, 9, 42. [Google Scholar] [CrossRef]
- Qiao, X.; Qin, X.; She, D.; Wang, R.; Zhang, X.; Zhang, L.; Zhang, Y. Mass Spectrometry-Based Tag and Its Application to High Efficient Peptide Analysis—A Review. Talanta 2014, 126, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Leitner, A.; Lindner, W. Chemistry Meets Proteomics: The Use of Chemical Tagging Reactions for MS-Based Proteomics. Proteomics 2006, 6, 5418–5434. [Google Scholar] [CrossRef]
- Leitner, A.; Lindner, W. Current Chemical Tagging Strategies for Proteome Analysis by Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 813, 1–26. [Google Scholar] [CrossRef]
- Miyagi, M.; Nakao, M.; Nakazawa, T.; Kato, I.; Tsunasawa, S. A Novel Derivatization Method with 5-Bromo-Nicotinic Acid N-Hydroxysuccinimide for Determination of the Amino Acid Sequences of Peptides. Rapid Commun. Mass Spectrom. 1998, 12, 603–608. [Google Scholar] [CrossRef]
- An, M.; Zou, X.; Wang, Q.; Zhao, X.; Wu, J.; Xu, M.; Shen, H.; Xiao, X.; He, D.; Ji, J.; et al. High-Confidence De Novo Peptide Sequencing Using Positive Charge Derivatization and Tandem MS Spectra Merging. Anal. Chem. 2013, 85, 4530–4537. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Shi, X.; Feng, Y.; Kent, K.C.; Li, L. Improving Data Quality and Preserving HCD-Generated Reporter Ions with EThcD for Isobaric Tag-Based Quantitative Proteomics and Proteome-Wide PTM Studies. Anal. Chim. Acta 2017, 968, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, F.; Liu, Z.; Qin, H.; Song, C.; Huang, J.; Bian, Y.; Wei, X.; Donga, J.; Zou, H. Five-Plex Isotope Dimethyl Labeling for Quantitative Proteomics. Chem. Commun. 2014, 50, 1708–1710. [Google Scholar] [CrossRef]
- Oldekop, M.L.; Herodes, K.; Rebane, R. Comparison of Amino Acid Derivatization Reagents for Liquid Chromatography Atmospheric Pressure Chemical Ionization Mass Spectrometric Analysis of Seven Amino Acids in Tea Extract. Int. J. Mass Spectrom. 2017, 421, 189–195. [Google Scholar] [CrossRef]
- Rebane, R.; Rodima, T.; Kütt, A.; Herodes, K. Development of Amino Acid Derivatization Reagents for Liquid Chromatography Electrospray Ionization Mass Spectrometric Analysis and Ionization Efficiency Measurements. J. Chromatogr. A 2015, 1390, 62–70. [Google Scholar] [CrossRef]
- Pohlentz, G.; Schlemm, S.; Klima, B.; Egge, H. Fast Atom Bombardment Mass Spectrometry of N-Acetylated Neoglycolipids of the 1-Deoxy-1-Phosphatidylethanolaminolactitol-Type. Chem. Phys. Lipids 1994, 70, 83–94. [Google Scholar] [CrossRef]
- Tsai, P.K.; Dell, A.; Ballou, C.E. Characterization of Acetylated and Acetolyzed Glycoprotein High-Mannose Core Oligosaccharides by Fast-Atom-Bombardment Mass Spectroscopy. Proc. Natl. Acad. Sci. USA 1986, 83, 4119–4123. [Google Scholar] [CrossRef]
- Claeys, M.; Claereboudt, J.; Uia, A. Fast Atom Bombardment Ionization in Mass Spectrometry, 3rd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; ISBN 9780128032244. [Google Scholar]
- Yang, H.-J.; Hu, X.-Y.; Chen, Y.-Z. Amino Acid Identification and Sequence Analysis of Peptides by Reaction Mass Spectrometry. Chin. J. Chem. 1993, 11, 540–549. [Google Scholar] [CrossRef]
- Paulson, J.; Lindberg, C. Increasing Thermospray Response for Cortisol by Derivatization. J. Chromatogr. A 1991, 554, 149–154. [Google Scholar] [CrossRef]
- Cho, K.; Kang, W.; Choi, Y.; Kim, W.; Pyo, K. Effects of Peptide Acetylation and Dimethylation on Electrospray Ionization Efficiency. J. Mass Spectrom. 2016, 51, 105–110. [Google Scholar] [CrossRef]
- Zappacosta, F.; Wagner, C.D.; Della Pietra, A.; Gerhart, S.V.; Keenan, K.; Korenchuck, S.; Quinn, C.J.; Barbash, O.; McCabe, M.T.; Annan, R.S. A Chemical Acetylation-Based Mass Spectrometry Platform for Histone Methylation Profiling. Mol. Cell. Proteomics 2021, 20, 100067. [Google Scholar] [CrossRef]
- Kuchibhotla, B.; Kola, S.R.; Medicherla, J.V.; Cherukuvada, S.V.; Dhople, V.M.; Nalam, M.R. Combinatorial Labeling Method for Improving Peptide Fragmentation in Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1216–1226. [Google Scholar] [CrossRef]
- Samgina, T.Y.; Kovalev, S.V.; Gorshkov, V.A.; Artemenko, K.A.; Poljakov, N.B.; Lebedev, A.T. N-Terminal Tagging Strategy for De Novo Sequencing of Short Peptides by ESI-MS/MS and MALDI-MS/MS. J. Am. Soc. Mass Spectrom. 2010, 21, 104–111. [Google Scholar] [CrossRef]
- Straube, E.; Dekant, W.; Völkel, W. Enhanced Sensitivity for the Determination of Ambiphilic Polyaromatic Amines by LC-MS/MS after Acetylation. J. Chromatogr. A 2005, 1067, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Jianying, Z.; Kodama, H. Liquid Chromatographic-Mass Spectrometric Analysis of N-Acetylamino Acids in Human Urine. J. Chromatogr. B 1994, 657, 15–21. [Google Scholar] [CrossRef]
- Chandra, D.; Gayathri, P.; Vats, M.; Nagaraj, R.; Ray, M.K. Mass Spectral Analysis of Acetylated Peptides: Implications in Proteomics. Eur. J. Mass Spectrom. 2019, 26, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G.; Young, A.B.; Bleiholder, C. Scrambling of Sequence Information in Collision-Induced Dissociation of Peptides. J. Am. Chem. Soc. 2006, 128, 10364–10365. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G. To b or Not to b: The Ongoing Saga of Peptide b Ions. Mass Spectrom. Rev. 2009, 28, 640–654. [Google Scholar] [CrossRef]
- Yaqüe, J.; Paradela, A.; Ramos, M.; Ogueta, S.; Marina, A.; Barahona, F.; López de Castro, J.A.; Vázquez, J. Peptide Rearrangement during Quadrupole Ion Trap Fragmentation: Added Complexity to MS/MS Spectra. Anal. Chem. 2003, 75, 1524–1535. [Google Scholar] [CrossRef]
- Harrison, A.G. Peptide Sequence Scrambling Through Cyclization of B5 Ions. J. Am. Soc. Mass Spectrom. 2008, 19, 1776–1780. [Google Scholar] [CrossRef] [PubMed]
- O’Hair, R.A.J.; Reid, G.E. Derivatization of Protonated Peptides via Gas Phase Ion-Molecule Reactions with Acetone. J. Am. Soc. Mass Spectrom. 2000, 11, 244–256. [Google Scholar] [CrossRef]
- Medzihradszky, K.F.; Chalkley, R.J. Lessons in De Novo Peptide Sequencing by Tandem Mass Spectrometry. Mass Spectrom. Rev. 2015, 34, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Hiserodt, R.D.; Brown, S.M.; Swijter, D.F.H.; Hawkins, N.; Mussinan, C.J. A Study of B1+H2O and B1-Ions in the Product Ion Spectra of Dipeptides Containing N-Terminal Basic Amino Acid Residues. J. Am. Soc. Mass Spectrom. 2007, 18, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Dabir, A.; Misal, S.A.; Tang, H.; Radivojac, P.; Reilly, J.P. The Impact of Amidination on Peptide Fragmentation and Identification in Shotgun Proteomics. J. Proteome Res. 2016, 15, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Thorne, G.C.; Gaskell, S.J.; Gross, M.L. Elucidation of Some Fragmentations of Small Peptides Using Sequential Mass Spectrometry on a Hybrid Instrument. Rapid Commun. Mass Spectrom. 1989, 3, 217–221. [Google Scholar] [CrossRef]
- Thorne, G.C.; Ballard, K.D.; Gaskell, S.J. Metastable Decomposition of Peptide [M + H]+ Ions via Rearrangement Involving Loss of the C-Terminal Amino Acid Residue. J. Am. Soc. Mass Spectrom. 1990, 1, 249–257. [Google Scholar] [CrossRef]
- Newton, K.A.; McLuckey, S.A. Generation and Manipulation of Sodium Cationized Peptides in the Gas Phase. J. Am. Soc. Mass Spectrom. 2004, 15, 607–615. [Google Scholar] [CrossRef]
- Biemann, K.; Martin, S.A. Mass Spectrometric Determination of the Amino Acid Sequence of Peptides and Proteins. Mass Spectrom. Rev. 1987, 6, 1–75. [Google Scholar] [CrossRef]
- Hunter, E.P.L.; Lias, S.G. Evaluate Gas Phase Basicities and Proton Affinity of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]
- Schneider, R.P.; Lynch, M.J.; Ericson, J.F.; Fouda, H.G. Electrospray Ionization Mass Spectrometry of Semduramicin and Other Polyether Ionophores. Anal. Chem. 1991, 63, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhang, Z.P.; Karnes, H.T. Sensitivity Enhancement in Liquid Chromatography/Atmospheric Pressure Ionization Mass Spectrometry Using Derivatization and Mobile Phase Additives. J. Chromatogr. B 2005, 825, 98–110. [Google Scholar] [CrossRef]
- Fenn, J.B. Ion Formation from Charged Droplets: Roles of Geometry, Energy, and Time. J. Am. Soc. Mass Spectrom. 1993, 4, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Iribarne, J.V.; Dziedzic, P.J.; Thomson, B.A. Atmospheric Pressure Ion Evaporation-Mass Spectrometry. Int. J. Mass Spectrom. Ion Phys. 1983, 50, 331–347. [Google Scholar] [CrossRef]
- Cech, N.B.; Enke, C.G. Practical Implications of Some Recent Studies in Electrospray Ionization Fundamentals. Mass Spectrom. Rev. 2001, 20, 362–387. [Google Scholar] [CrossRef]
- Sun, B.; Liu, Z.; Liu, J.; Zhao, S.; Wang, L.; Wang, F. The Utility of Proteases in Proteomics, from Sequence Profiling to Structure and Function Analysis. Proteomics 2022, 23, 2200132. [Google Scholar] [CrossRef]
- Kloudová, B.; Strmeň, T.; Vrkoslav, V.; Chára, Z.; Pačes, O.; Cvačka, J. Gas Dynamic Virtual Nozzle Sprayer for an Introduction of Liquid Samples in Atmospheric Pressure Ionization Mass Spectrometry. Anal. Chem. 2022, 95, 4196–4203. [Google Scholar] [CrossRef]
- Strmeň, T.; Vrkoslav, V.; Bosáková, Z.; Cvačka, J. Atmospheric Pressure Chemical Ionization Mass Spectrometry at Low Flow Rates: Importance of Ion Source Housing. Rapid Commun. Mass Spectrom. 2020, 34, e8722. [Google Scholar] [CrossRef]
- Vrkoslav, V.; Rumlová, B.; Strmeň, T.; Nekvasilová, P.; Šulc, M.; Cvačka, J. Applicability of Low-Flow Atmospheric Pressure Chemical Ionization and Photoionization Mass Spectrometry with a Microfabricated Nebulizer for Neutral Lipids. Rapid Commun. Mass Spectrom. 2018, 32, 639–648. [Google Scholar] [CrossRef]
- Strmeň, T.; Vrkoslav, V.; Pačes, O.; Cvačka, J. Evaluation of an Ion Source with a Tubular Nebulizer for Microflow Atmospheric Pressure Chemical Ionization. Mon. Für Chem. 2018, 149, 987–994. [Google Scholar] [CrossRef]
- Erde, J.; Loo, R.R.O.; Loo, J.A. Improving Proteome Coverage and Sample Recovery with Enhanced FASP (EFASP) for Quantitative Proteomic Experiments. Methods Mol. Biol. 2017, 1550, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gostian, A.O.; Hüttenbrink, K.B.; Luers, J.C.; Anagiotos, A.; Beutner, D. Probability-Based Protein Identification by Searching Sequence Databases Using Mass Spectrometry Data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Bian, Y.; Gao, C.; Kuster, B. On the Potential of Micro-Flow LC-MS/MS in Proteomics. Expert Rev. Proteom. 2022, 19, 153–164. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion Source | Protease | Sequence Coverage [%] | ||
|---|---|---|---|---|
| Acetylation at the Protein Level | Acetylation at the Peptides Level | No Acetylation | ||
| APCI | Chymotrypsin | 30 | 25 | 21 |
| Glu-C | 25 | 28 | 22 | |
| APPI | Chymotrypsin | 34 | 43 | 22 |
| Glu-C | 54 | 62 | 42 | |
| ESI | Chymotrypsin | 40 | 40 | 37 |
| Glu-C | 39 | 44 | 37 | |
| Protease | Type of Peptides | Number of Peptides | ||
|---|---|---|---|---|
| Acetylation at the Protein Level | Acetylation at the Peptides Level | No Acetylation | ||
| Chymotrypsin | All | 41 | 36 | 29 |
| Acetylated methyl esters | 1 | 1 | 0 | |
| Long peptides * | 7 | 8 | 7 | |
| Sodium adducts | 0 | 0 | 0 | |
| Glu-C | All | 22 | 23 | 21 |
| Acetylated methyl esters | 2 | 7 | 0 | |
| Long peptides * | 11 | 16 | 13 | |
| Sodium adducts | 0 | 0 | 0 | |
| Protease | Types of Peptide | Number of Peptides | ||
|---|---|---|---|---|
| Acetylation at the Protein Level | Acetylation at the Peptides Level | No Acetylation | ||
| Chymotrypsin | All | 27 | 34 | 30 |
| Acetylated methyl esters | 2 | 0 | 0 | |
| Long peptides * | 17 | 19 | 6 | |
| Sodium adducts | 14 | 16 | 0 | |
| Glu-C | All | 44 | 47 | 36 |
| Acetylated methyl esters | 2 | 10 | 0 | |
| Long peptides * | 36 | 39 | 28 | |
| Sodium adducts | 21 | 26 | 4 | |
| Protease | Type of Peptides | Number of Peptides | ||
|---|---|---|---|---|
| Acetylation at the Protein Level | Acetylation at the Peptides Level | No Acetylation | ||
| Chymotrypsin | All | 27 | 40 | 35 |
| Acetylated methyl esters | 3 | 4 | 0 | |
| Long peptides * | 20 | 23 | 23 | |
| Sodium adducts | 3 | 1 | 2 | |
| Singly charged | 15 | 28 | 11 | |
| Doubly charged | 10 | 10 | 15 | |
| Triply charged | 2 | 2 | 8 | |
| Quadruply charged | 0 | 0 | 1 | |
| Glu-C | All | 25 | 34 | 30 |
| Acetylated methyl esters | 2 | 6 | 0 | |
| Long peptides * | 21 | 24 | 23 | |
| Sodium adducts | 0 | 0 | 3 | |
| Singly charged | 15 | 20 | 7 | |
| Doubly charged | 10 | 14 | 18 | |
| Triply charged | 0 | 0 | 5 | |
| Quadruply charged | 0 | 0 | 0 |
| Protease | Sequence Coverage [%] | |||
|---|---|---|---|---|
| Acetylated at the Protein Level * | Acetylated at the Peptides Level * | |||
| ESI (NAC) + APCI | ESI (NAC) + APPI | ESI (NAC) + APCI | ESI (NAC) + APPI | |
| Chymotrypsin | 49 | 56 | 47 | 54 |
| Glu-C | 48 | 70 | 49 | 71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedláčková, S.; Hubálek, M.; Vrkoslav, V.; Blechová, M.; Kozlík, P.; Cvačka, J. Positive Effect of Acetylation on Proteomic Analysis Based on Liquid Chromatography with Atmospheric Pressure Chemical Ionization and Photoionization Mass Spectrometry. Molecules 2023, 28, 3711. https://doi.org/10.3390/molecules28093711
Sedláčková S, Hubálek M, Vrkoslav V, Blechová M, Kozlík P, Cvačka J. Positive Effect of Acetylation on Proteomic Analysis Based on Liquid Chromatography with Atmospheric Pressure Chemical Ionization and Photoionization Mass Spectrometry. Molecules. 2023; 28(9):3711. https://doi.org/10.3390/molecules28093711
Chicago/Turabian StyleSedláčková, Simona, Martin Hubálek, Vladimír Vrkoslav, Miroslava Blechová, Petr Kozlík, and Josef Cvačka. 2023. "Positive Effect of Acetylation on Proteomic Analysis Based on Liquid Chromatography with Atmospheric Pressure Chemical Ionization and Photoionization Mass Spectrometry" Molecules 28, no. 9: 3711. https://doi.org/10.3390/molecules28093711