
On the Many-Body Expansion of an Interaction Energy of Some Supramolecular Halogen-Containing Capsules


Institute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovsky Square, 16206 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(15), 4431; https://doi.org/10.3390/molecules26154431
Submission received: 25 June 2021
/
Revised: 18 July 2021
/
Accepted: 19 July 2021
/
Published: 22 July 2021
Abstract
:A tetramer model was investigated of a remarkably stable iodine-containing supramolecular capsule that was most recently characterized by other authors, who described emergent features of the capsule’s formation. In an attempt to address the surprising fact that no strong pair-wise interactions between any of the respective components were experimentally detected in condensed phases, the DFT (density-functional theory) computational model was used to decompose the total stabilization energy as a sum of two-, three- and four-body contributions. This model considers complexes formed between either iodine or bromine and the crucial D4d-symmetric form of octaaryl macrocyclic compound cyclo[8](1,3-(4,6-dimethyl)benzene that is surrounded by arenes of a suitable size, namely, either corannulene or coronene. A significant enthalpic gain associated with the formation of investigated tetramers was revealed. Furthermore, it is shown that the total stabilization of these complexes is dominated by binary interactions. Based on these findings, comments are made regarding the experimentally observed behavior of related multicomponent mixtures.
1. Introduction
The molecular self-assembly is one of the most important directions in supramolecular chemistry [1], and a number of applications of synthetic self-assembled architectures were reported (see recent reviews [2,3,4,5,6] and references cited therein). Many multicomponent self-assembled systems were prepared, which were surveyed by Yang et al. [7]. Their association is known to be driven by direct interactions between pairs of the components. Most recently, however, Yang et al. [7] obtained a remarkably stable multicomponent self-assembled capsule (see below), while no pair-wise interactions between its respective subunits were apparent in solution and solid states. This is clearly an important finding, whose significance goes beyond synthetic organic chemistry. It is relevant to crystal engineering [8] and to molecular recognition through non-covalent interactions in general [9], in addition to its possible applications in a preparation of materials with desirable properties, as already described in reference [7]. The structural model of related multicomponent aggregates was thus investigated here. Specifically, high-level quantum chemical calculations were applied in order to estimate the respective contributions to the total stabilization of several four-component systems. Those systems consist of a “shell” formed by two arenes (either corannulenes or coronenes) around the crucial macrocycle of approximately D4d-symmetric cyclo[8](1,3-(4,6-dimethyl)benzene [10,11] that is abbreviated as CDMB in the following, and of a “core” containing either molecular iodine or molecular bromine (I2 or Br2). Answers are sought to the following three main questions: (1) How big is the enthalpic gain associated with a formation of the CDMB-containing tetramers? (2) What is the breakdown into two-, three-, and four-body contributions of the total stabilization of those complexes? (3) Are there vast differences in the enthalpic stabilization predicted for different tetramers (containing CDMB together with either two corannulenes or two coronenes and either one I2 or one Br2 molecule; throughout the article, acronyms Cora and COR are used for corannulene and coronene, respectively)? The presented DFT (density-functional theory) computational results are expected to be useful in exploring the relationship between intermolecular interactions and stability of molecular containers. In particular, they provide context for “emergent behavior” of the iodine-containing capsule most recently described by Yang et al. [7], and thus they contribute to the understanding of factors that govern the self-assembly mechanisms (see the important review article [12]).
2. Results and Discussion
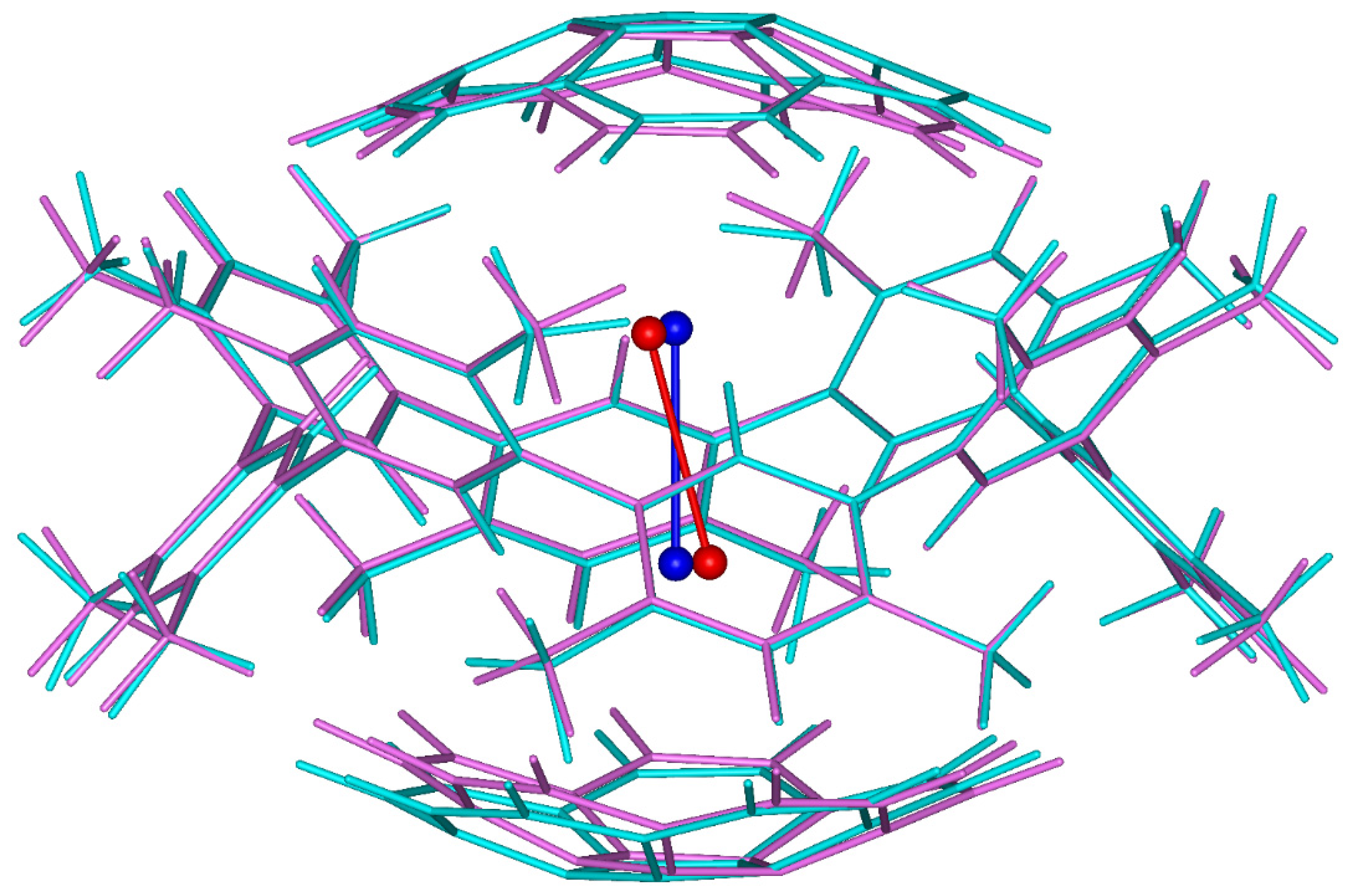
The tetramer formed by one CDMB molecule with iodine inside and surrounded by two coroannulenes is shown in Figure 1 (see also Scheme 1). Its initial geometry was clipped out from the X-ray diffraction (XRD) structure of a cyclohexane solvate of the iodine-containing capsule [7]. In that structure, whose CCSD refcode is IKEZUW, molecular iodine can be seen to be parallel to the C4 rotation axis of neighboring CDMB units. This symmetry was broken in the course of the DFT optimization (the methodology is detailed in Part 3). However, the angle is small between initial and final I2 bond vectors, namely, it amounts to 15°. In addition, orientations of the macrocycle and corannulene units are quite similar in the XRD and DFT geometries (see Figure 1).
Table 1 presents the decomposition of many-body contributions to the DFT interaction energy of the tetramer supermolecule, which were obtained on the basis of Equations (1) and (2) that are given in the Materials and Methods section. The value is completely dominated by the following terms (their sum amounts to 98.9% of the total interaction energy): the dimers of CDMB and either corannulene (62.6%), the dimers of iodine and either corannulene (22.1%), and the CDMB…I2 dimer (14.2%). These values were checked against their counterparts obtained from the RI-MP2/def2-TZVP energies (the resolution-of-the-identity integral approximation to the second-order Møller–Plesset method combined with an application of the triple-zeta valence plus polarization basis set; see Part 3 for details) computed for the same DFT geometry. The MP2-based results are qualitatively similar: in this case the value almost exactly (within 1%) amounts to a sum of contributions from dimers of CDMB and corannulenes (58.0%), of iodine and corannulenes (28.3%), and of iodine and CDMB (13.0%), while all the remaining terms are small (see the Supplementary Materials Table S1). Hence, there is practically no cooperativity (in its classical definition [13]) in the enthalpic stabilization of this tetramer. It is an important finding in the context of experiments [7], which did not reveal any strong binary interactions that would lead to a formation of related dimeric complex(es) in condensed phases. It thus seems that in mixtures containing CDMB, corannulene and iodine, cyclohexane solvent molecules played a decisive role in the emergent behavior of this system that was most recently reported by Yang et al. [7] (its components did not crystallize or associate in solution in pair-wise fashion, but together they formed a highly stable capsule). At this point, it is worth bringing an example of the ethanol tetramer, in which many-body effects are crucial for its enthalpic stabilization. Specifically, the trimers account for 21.4% of the value (details are provided in the Supplementary Materials, Table S3).
The DFT calculations were also used to search the potential-energy surface of all constituting dimers and trimers to try to locate their global minima and subsequently evaluate thermodynamic parameters. Unfortunately, no minimum was found in the case of CDMB…Cora…I2 trimer. Its lowest-energy structure, which is a transition state, is considered below anyway. Results are summarized in Table 2, wherein data denoted as refer to the CP corrected interaction energy, while refers to the Gibbs free energy of formation estimated at T = 298.15 K using a crude model [14]. Of course, it is not possible to immediately compare the interaction energy of isolated dimers with the corresponding of dimers embedded in the tetramer from Table 1, because different geometries and counterpoise procedures were adopted in the respective calculations. Still, it is worth noting that the enthalpic gain during the tetramer formation is high, as exceeds −300 kJ/mol, while the stabilization conferred by CDMB…Cora dimers is around −100 kJ/mol, for Cora…I2 is around −40 kJ/mol, etc. An inspection of values reveals that the stabilization of trimers is approximately additive (for example, of the CDMB…Cora dimer is −96.3 kJ/mol that amounts to 49.1% of the interaction energy of −196.1 kJ/mol of the trimer formed by one CDMB and two corannulenes). Interestingly, a spontaneous formation of all the dimers and trimers at room temperature is predicted on the basis of estimated data. This can be contrasted with the situation of ethanol tetramer, whose is +383 kJ/mol due to a large entropic penalty vastly surpassing the enthalpic stabilization (see the Supplementary Materials, Table S4, for details). Results of the present calculations show that there is a substantial intrinsic tendency of all three components of the capsule (CDMB, corannulene, and I2) to associate despite an absence of highly stabilizing contacts (like salt bridges or strong hydrogen bonds) in the studied complexes. This tendency is yet another indication that cyclohexane molecules are crucial for the emergent behavior of the investigated system [7].
It could be of interest to compare the interaction energies obtained for the aforementioned cluster to its several alternatives, which feature different “shell” and/or “core” fragments of theoretical models of the capsule. Table 3 summarizes results for the four tetramers considered here. From among them, the highest binding energy of ca. −288 kJ/mol is found for the structure already described. This binding energy is followed by a value, which is lower by ca. 18 kJ/mol, that pertains to the tetramer containing molecular bromine instead of iodine. Importantly, Yang et al. [7] presented a preliminary characterization of bromine capture from mixtures containing CDMB and corannulene, and so it is likely that a related self-assembled system will be prepared. The binding energies of coronene-containing tetramers are further lowered by additional ca. 13 and 14 kJ/mol for systems with bromine and iodine, respectively (see Table 3). These differences are relatively small, and hence it appears that also coronenes might form a shell part of complexes with CDMB and a host molecule. The decomposition of a total CP interaction energy obtained for the three modified clusters is shown in Table 4. In the same way as in the cluster presented in Table 1, predominant contributions to come from neighboring dimers.
3. Computational Methods
The following monomers and their point group symmetry (in parentheses) were considered: I2 (D∞h); Br2 (D∞h); CDMB (D4d); corannulene, abbreviated as Cora (C5v); coronene, abbreviated as COR (D6h); and ethanol (Cs). The isolated dimers and trimers were: CDMB…Cora; CDMB…I2; Cora…Cora; Cora…I2; Cora…CDMB…Cora; Cora…CDMB…I2; and Cora…I2…Cora, and were all of C1 symmetry. Of C1 symmetry were also tetramers comprising: CDMB, two Cora molecules, and I2; CDMB, two COR molecules, and I2; CDMB, two Cora molecules, and Br2; and CDMB, two COR molecules, and Br2. The investigated ethanol tetramer was of S4 symmetry (it is a cyclic structure comprising monomers in gauche configuration [15]). All these structures were treated at the B3LYP-D3/6-311G** level of quantum chemical theory, that is, by combining the standard Becke’s three-parameter exchange and the Lee–Yang–Parr correlation DFT functionals together with an unmodified D3 empirical dispersion-correction [16] and with the standard all-electron 6-311G** basis set. Their geometries were fully optimized, and their harmonic vibrational analysis was performed to obtain values of the zero-point energy, the vibrational thermal energy, and the entropy of the respective components in order to estimate in a routine way [14] the Gibbs free energy change at 298.15 K accompanying the formation of the aforementioned dimers and trimers that is denoted simply as . Subsequently, the deformation energy, , of the monomers was obtained as the energy difference between the monomer embedded in an investigated complex and of an isolated monomer. An influence of scalar relativistic effects upon the interaction energies, , was approximated at the B3LYP-D3 level while using the Douglas–Kroll–Hess approach combined with the triple-zeta valence plus polarization basis sets of Jorge et al. [17]. The and data were added to the total counterpoise-corrected (CP) interaction energy [18] (see right below), and to the related difference of zero-point energies, , to arrive at a value of the binding energy, , of investigated tetramers. The CP complexation energies were computed using the site–site function counterpoise method [19]. In the following, respective components of a tetramer are denoted by integers from the set and the indices go over them. Additionally, the superscript is always used to indicate that in this method the basis functions are at all nuclei of an investigated tetramer (the situation could be different if other schemes were used, see reference [20] for a recent discussion). Thus, for instance, symbol designates the energy of a full tetramer; is the energy of a first unit with ghost nuclei in the positions of units , , and ; is the energy of the dimer comprising units and , and ghost nuclei of units and , etc. Based on an expansion of the total energy of a cluster containing particles into one-particle, two-particle, three-particle, … etc. energies [21], the following two equations for the CP interaction energy are formed:
and
where
and
The shorthand notation in “(monomer)x” subscript is used for the corresponding monomers, “(two-body)y” for dimers together with relevant monomers, and “(three-body)z” for trimers together with relevant dimers and monomers. The inclusion–exclusion formula [22] was applied in Equation (2) in order to avoid double counting of the respective contributions, as the presented procedure requires only 15 energies to be computed to obtain all terms of Equations (1) and (2) in a general case. It should be noted that this number is lowered if a tetramer is symmetric. Specifically, the number of unique energies is five in the case of the S4-symmetric ethanol tetramer. They are together with for example , , , and (the energy of one of the monomers, one of the neighbor dimers, one of the diagonal dimers, and one of the trimers, respectively).
In addition to the aforementioned calculations on target systems, the B3LYP-D3/6-311G** interaction energies were obtained for a small set of halogen-containing dimers (see the Supporting Materials, Table S2 and Figure S1), whose highly accurate geometries obtained by Hobza and his coworkers [23] were taken from the BEGDB database [24], so as to confirm that this computational approach provides qualitatively correct results for various types of intermolecular interactions involving iodine and bromine sites. Computations were carried out using the Gaussian 16 program package [25]. Default settings were applied except for the interaction energy calculations, which were performed with a tighter integration grid, namely, with “Grid = SuperFine” option of “Integral” parameter. All the B3LYP-D3/6-311G** optimized geometries and total energies are provided in the Supporting Materials.
The B3LYP-D3/6-311G** geometry of the tetramer comprising CDMB, two Cora molecules, and I2 was also employed to obtain the interaction energies at the RI-MP2/def2-TZVP level, namely, by using the RI integral approximation [26] and the corresponding orbital [27] and auxiliary [28] basis sets in version 7.1 of the Turbomole software [29].
4. Conclusions
Results of the present B3LYP-D3/6-311G** calculations show a strong enthalpic stabilization of molecular iodine inside the CDMB macrocycle surrounded by coronenes, which is in line with the suggestion that those components are in a thermodynamic minimum under the conditions described in reference [7]. The many-body decomposition of the total interaction energy of the tetramer model reveals only negligible contributions from three- and four-body terms. This is an indirect indication of cyclohexane solvent leading to the emergent behavior of the multicomponent mixtures, which was observed experimentally [7]. Tetramers with the changed composition (iodine replaced with bromine, corannulenes replaced with coronenes) are predicted to be considerably stable, too, and thus the corresponding systems are expected to also form supramolecular capsules.
Supplementary Materials
The following are available online. “SI.PDF” file containing comparison of the RI-MP2 and DFT results (Table S1); description of the test set of interaction energies (Table S2 and Figure S1); and additional data for the ethanol tetramer (Tables S3 and S4). Optimized coordinates and their total energies in XYZ format files, which have “xyz” suffix and self-explanatory names: I2, Br2, CDMB, Cora, COR, ethanol, CDMB-Cora, CDMB-I2, Cora-Cora, Cora-I2, Cora-CDMB-Cora, Cora-CDMB-I2, Cora-I2-Cora, Cora-CDMB-I2-Cora, Cora-CDMB-Br2-Cora, COR-CDMB-I2-COR, COR-CDMB-Br2-COR, and ethanol_tetramer.
Author Contributions
Conceptualization, J.C. and J.B.; investigation, writing, J.C.; validation, funding acquisition, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
Ministry of Education, Youth and Sports of the Czech Republic project LTAUSA18011.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and in the supplementary materials.
Acknowledgments
Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures; and by the ELIXIR-CZ project (LM2015047), part of the international ELIXIR infrastructure. The support provided by the Czech Academy of Sciences within the programme AV21 Strategy: RP10-Molecules and Materials for Life is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Wang, H.; Wang, K.; Xu, Y.; Wu, W.; Chen, S.; Hart, M.; Wojtas, L.; Zhou, L.-P.; Gan, L.; Yan, X.; et al. Hierarchical Self-Assembly of Nanowires on the Surface by Metallo-Supramolecular Truncated Cuboctahedra. J. Am. Chem. Soc. 2021, 143, 5826–5835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ronson, T.K.; Zou, Y.-Q.; Nitschke, J.R. Metal–organic cages for molecular separations. Nat. Rev. Chem. 2021, 5, 168–182. [Google Scholar] [CrossRef]
- Percástegui, E.G.; Ronson, T.K.; Nitschke, J.R. Design and Applications of Water-Soluble Coordination Cages. Chem. Rev. 2020, 120, 13480–13544. [Google Scholar] [CrossRef]
- Gao, W.-X.; Feng, H.-J.; Guo, B.-B.; Lu, Y.; Jin, G.-X. Coordination-Directed Construction of Molecular Links. Chem. Rev. 2020, 120, 6288–6325. [Google Scholar] [CrossRef]
- Pilgrim, B.S.; Champness, N.R. Metal-Organic Frameworks and Metal-Organic Cages—A Perspective. ChemPlusChem 2020, 85, 1842–1856. [Google Scholar] [CrossRef]
- Dong, J.; Liu, Y.; Cui, Y. Supramolecular Chirality in Metal–Organic Complexes. Acc. Chem. Res. 2021, 54, 194–206. [Google Scholar] [CrossRef]
- Yang, Y.-D.; Chen, X.-L.; Sessler, J.L.; Gong, H.-Y. Emergent Self-Assembly of a Multicomponent Capsule via Iodine Capture. J. Am. Chem. Soc. 2021, 143, 2315–2324. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Bučar, D.-K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
- Persch, E.; Dumele, O.; Diederich, F. Molecular Recognition in Chemical and Biological Systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef]
- Yang, Y.-D.; Gong, H.-Y. Thermally activated isomeric all-hydrocarbon molecular receptors for fullerene separation. Chem. Commun. 2019, 55, 3701–3704. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-D.; Ji, X.; Lu, Z.-H.; Yang, J.; Gao, C.; Tang, B.; Sessler, J.L.; Gong, H.-Y. Time dependent solid-state molecular motion and colour tuning of host-guest systems by organic solvents. Nat. Commun. 2020, 11, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Stoddart, J.F. Emergent behavior in nanoconfined molecular containers. Chem 2021, 7, 919–947. [Google Scholar] [CrossRef]
- Koehler, J.E.H.; Saenger, W. Ab Initio Calculations and Many Body Analysis of the Water Tetramer. In Interactions of Water in Ionic and Nonionic Hydrates; Kleeberg, H., Ed.; Springer: Berlin/Heidelberg, Germany, 1987; pp. 145–146. [Google Scholar]
- Irikura, K.K.; National Institute of Standards and Technology. Using the Output File from a Gaussian Frequency Calculation to Compute Ideal-Gas Thermodynamic Functions. Available online: https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas/ (accessed on 25 June 2021).
- Šoltésová, M.; Benda, L.; Peksa, M.; Czernek, J.; Lang, J. Determination of Size of Molecular Clusters of Ethanol by Means of NMR Diffusometry and Hydrodynamic Calculations. J. Phys. Chem. B 2014, 118, 6864–6874. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorge, F.E.; Neto, A.C.; Camiletti, G.G.; Machado, S.F. Contracted Gaussian basis sets for Douglas–Kroll–Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 64108. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Wells, B.H.; Wilson, S. Van der Waals interaction potentials: Many-body basis set superposition effects. Chem. Phys. Lett. 1983, 101, 429–434. [Google Scholar] [CrossRef]
- Herbert, G.M. Fantasy versus reality in fragment-based quantum chemistry. J. Chem. Phys. 2019, 151, 170901. [Google Scholar] [CrossRef]
- Kaplan, I.G.; Santamaria, R.; Novaro, O. Non-additive forces in atomic clusters. Mol. Phys. 1995, 84, 105–114. [Google Scholar] [CrossRef]
- Bender, E.A.; Williamson, S.G. Foundations of Combinatorics with Applications, 1st ed.; Dover Publications: Mineola, NY, USA, 2006. [Google Scholar]
- Řezáč, J.; Riley, K.E.; Hobza, P. Benchmark Calculations of Noncovalent Interactions of Halogenated Molecules. J. Chem. Theory Comput. 2012, 8, 4285–4292. [Google Scholar] [CrossRef]
- The Benchmark Energy & Geometry Database (BEGDB). Available online: http://www.begdb.org/ (accessed on 25 June 2021).
- Frish, M.J.; Trucks, J.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Hättig, C.; Hellweg, A.; Köhn, A. Distributed memory parallel implementation of energies and gradients for second-order Møller–Plesset perturbation theory with the resolution-of-the-identity approximation. Phys. Chem. Chem. Phys. 2006, 8, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design an assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Initial and final geometries of the tetramer discussed in the text. Color coding of the initial/final structures: blue/red for molecular iodine, cyan/magenta for the rest of the atoms.
Figure 1.
Initial and final geometries of the tetramer discussed in the text. Color coding of the initial/final structures: blue/red for molecular iodine, cyan/magenta for the rest of the atoms.

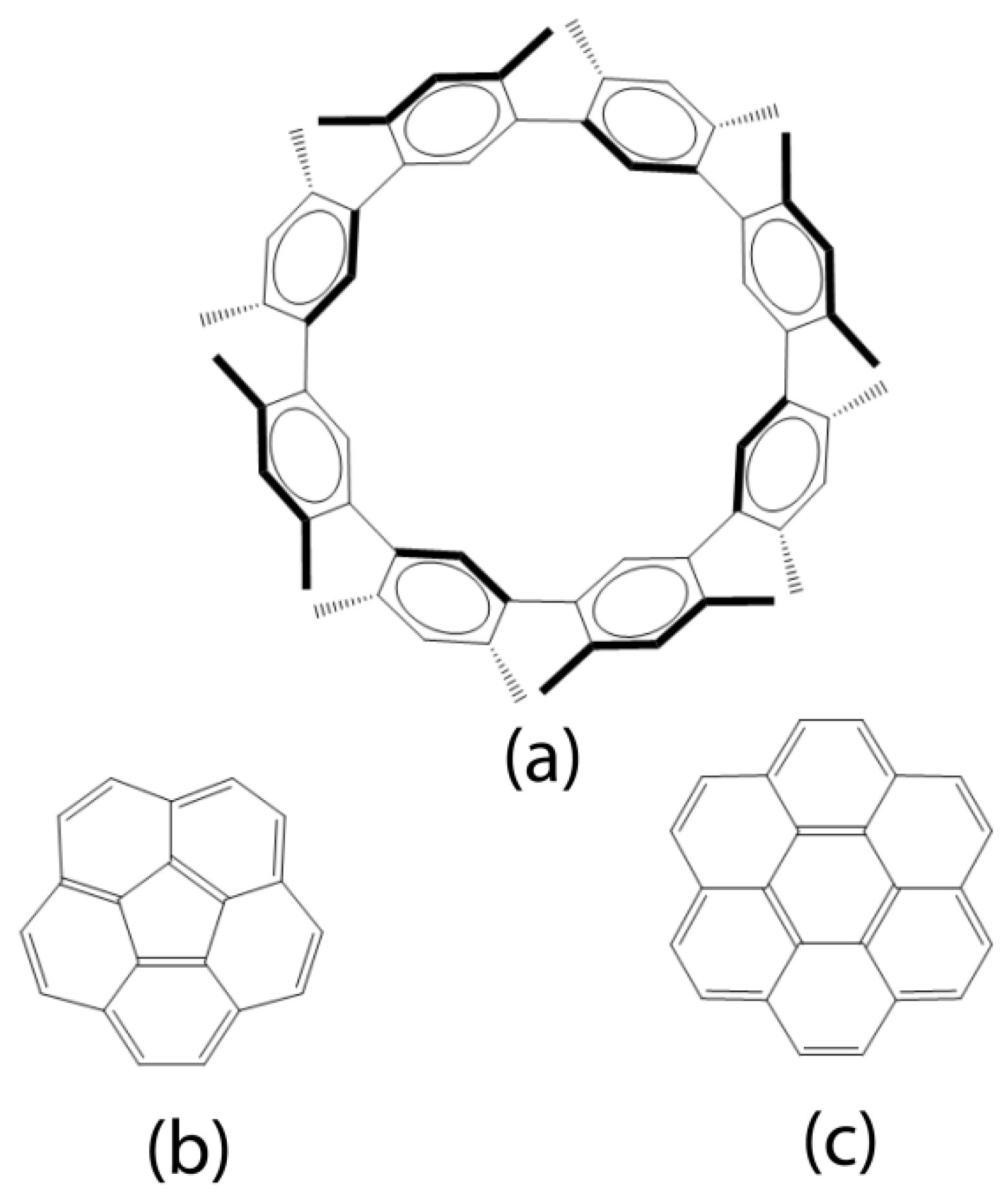
Scheme 1.
Schematic drawings of D4d-symmetric cyclo[8](1,3-(4,6-dimethyl)benzene, C5v-symmetric corranulene, and D6h-symmetric coronene in panels (a), (b), and (c), respectively.
Scheme 1.
Schematic drawings of D4d-symmetric cyclo[8](1,3-(4,6-dimethyl)benzene, C5v-symmetric corranulene, and D6h-symmetric coronene in panels (a), (b), and (c), respectively.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The breakdown of contributions to (in kJ/mol) defined in Equation (2).
| Interaction | ||
|---|---|---|
| Two-body | CDMB…Cora′ | −96.6 |
| CDMB…Cora″ | −96.8 | |
| CDMB…I2 | −44.0 | |
| Cora’…Cora″ | −1.1 | |
| Cora’…I2 | −34.1 | |
| Cora”…I2 | −34.0 | |
| Σ (dimers) | −306.5 | |
| Three-body | Cora’…CDMB…Cora” | −0.3 |
| Cora′…CDMB…I2 | −2.6 | |
| CDMB…I2…Cora″ | −2.5 | |
| Cora′…I2…Cora″ | +2.8 | |
| Σ (trimers) | −2.6 | |
| Four-body | Cora’…CDMB…I2…Cora” | +0.5 |
| Total | tetramer formation | −308.7 |
Table 2.
The energies (in kJ/mol) of the most energetically favorable structures as found during searches of the potential-energy surface. See the text for details.
Table 2.
The energies (in kJ/mol) of the most energetically favorable structures as found during searches of the potential-energy surface. See the text for details.
| Complex | ||
|---|---|---|
| CDMB…Cora | −96.3 | −35.3 |
| CDMB…I2 | −46.3 | −20.6 |
| Cora…Cora | −54.2 | −7.6 |
| Cora…I2 | −40.0 | −5.4 |
| Cora…CDMB…Cora | −196.1 | −70.7 |
| Cora…CDMB…I2 | −179.0 | −72.8 |
| Cora…I2…Cora | −91.6 | −6.8 |
Table 3.
The interaction energy contributions and the total binding energy of the investigated tetramers. All values are in kJ/mol.
Table 3.
The interaction energy contributions and the total binding energy of the investigated tetramers. All values are in kJ/mol.
| Term | System | |||
|---|---|---|---|---|
| CDMB(Cora)2I2 | CDMB(COR)2I2 | CDMB(Cora)2Br2 | CDMB(COR)2Br2 | |
| −308.7 | −273.5 | −293.6 | −278.6 | |
| 16.4 | 12.5 | 17.4 | 18.1 | |
| 6.7 | 9.4 | 10.6 | 4.6 | |
| −2.5 | −4.0 | −4.4 | −1.7 | |
| −288.1 | −255.6 | −270.0 | −257.5 | |
Table 4.
The breakdown of contributions to (in kJ/mol) of the three investigated tetramers.
| Interaction 1 | System | |||
|---|---|---|---|---|
| CDMB(COR)2I2 | CDMB(Cora)2Br2 | CDMB(COR)2Br2 | ||
| Two-body | A…B′ | −94.3 | −98.0 | −101.1 |
| A…B″ | −95.1 | −97.7 | −101.0 | |
| A…C | −39.9 | −35.1 | −31.8 | |
| B′…B″ | −0.6 | −1.4 | −1.0 | |
| B′…C | −23.1 | −29.3 | −21.4 | |
| B″…C | −23.2 | −29.3 | −21.3 | |
| Σ (dimers) | −276.2 | −290.7 | −277.6 | |
| Three-body | B′…A…B″ | −0.1 | −0.5 | −1.4 |
| B′…A…C | −0.3 | −2.0 | −1.3 | |
| A…C…B″ | −0.2 | −2.4 | −1.2 | |
| B′…C…B″ | +4.7 | +1.5 | +3.1 | |
| Σ (trimers) | +4.1 | −3.3 | −0.8 | |
| Four-body | B′…A…C…B″ | −1.4 | +0.4 | −0.2 |
| Total | tetramer formation | −273.5 | −293.6 | −278.6 |
1 Designated accordingly A…B′…B″…C.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Czernek, J.; Brus, J. On the Many-Body Expansion of an Interaction Energy of Some Supramolecular Halogen-Containing Capsules. Molecules 2021, 26, 4431. https://doi.org/10.3390/molecules26154431
AMA Style
Czernek J, Brus J. On the Many-Body Expansion of an Interaction Energy of Some Supramolecular Halogen-Containing Capsules. Molecules. 2021; 26(15):4431. https://doi.org/10.3390/molecules26154431
Chicago/Turabian StyleCzernek, Jiří, and Jiří Brus. 2021. "On the Many-Body Expansion of an Interaction Energy of Some Supramolecular Halogen-Containing Capsules" Molecules 26, no. 15: 4431. https://doi.org/10.3390/molecules26154431