
The Ubiquitination System within Bacterial Host–Pathogen Interactions


1
Department of Molecular Pathology and Biology, Faculty of Military Health Sciences, University of Defence, Trebesska 1575, 50001 Hradec Kralove, Czech Republic
2
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo namesti 542/2, 16000 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
Microorganisms 2021, 9(3), 638; https://doi.org/10.3390/microorganisms9030638
Submission received: 19 February 2021
/
Revised: 15 March 2021
/
Accepted: 16 March 2021
/
Published: 19 March 2021
(This article belongs to the Special Issue Interaction between Francisella Species and the Host Immune System)
Abstract
:Ubiquitination of proteins, like phosphorylation and acetylation, is an important regulatory aspect influencing numerous and various cell processes, such as immune response signaling and autophagy. The study of ubiquitination has become essential to learning about host–pathogen interactions, and a better understanding of the detailed mechanisms through which pathogens affect ubiquitination processes in host cell will contribute to vaccine development and effective treatment of diseases. Pathogenic bacteria (e.g., Salmonella enterica, Legionella pneumophila and Shigella flexneri) encode many effector proteins, such as deubiquitinating enzymes (DUBs), targeting the host ubiquitin machinery and thus disrupting pertinent ubiquitin-dependent anti-bacterial response. We focus here upon the host ubiquitination system as an integral unit, its interconnection with the regulation of inflammation and autophagy, and primarily while examining pathogens manipulating the host ubiquitination system. Many bacterial effector proteins have already been described as being translocated into the host cell, where they directly regulate host defense processes. Due to their importance in pathogenic bacteria progression within the host, they are regarded as virulence factors essential for bacterial evasion. However, in some cases (e.g., Francisella tularensis) the host ubiquitination system is influenced by bacterial infection, although the responsible bacterial effectors are still unknown.
1. Introduction
Competition between host defense mechanisms and pathogens’ effective tools has been observed since ancient times. The immune system of the host organism is regulated by complex metabolic and signaling pathways, including, but not limited to, a network of post-translational modifications. Should any of these important pathways be impaired, such as by efficient mechanisms of pathogens, the immune balance is disrupted and pathogens may become more successful. During their development, pathogenic organisms have acquired several mechanisms by which they can specifically influence the immune response of the host and thus escape their defense mechanisms and prevent themselves from destruction. Better understanding how pathogens affect the host immune system will facilitate the development of more effective therapeutic agents against diseases caused by pathogenic microorganisms. This review will be devoted to one of the post-translational modifications, ubiquitination, in connection with host–pathogen interaction. Ubiquitination is a crucial mechanism in numerous cell processes, such as protein degradation, innate immune signaling, protein–protein interactions, and others. Many intracellular pathogens intervene through the ubiquitination system into the host’s innate immune response. Ubiquitination, in addition to acetylation and phosphorylation, is becoming another massively studied modification in connection with host–pathogen interactions. The importance of investigating this post-translational modification in the context of the host–pathogen interaction is constantly increasing, particularly due to the availability of new and sensitive approaches for analyzing ubiquitination.
2. Ubiquitin System
Ubiquitination is a significant, reversible post-translational modification managing numerous cellular processes. This modification mediates not only the degradation of proteins but also ensures proper protein function, protein–protein interaction, and subcellular localization [1].
More than 500 proteins possess the ability to recognize ubiquitin (Ub) and Ub-like molecules. These proteins participate in connecting Ub to a specific substrate or its removal from the target molecule. The conjugation and deconjugation of Ub are broadly diverse and complicated processes that regulate many cellular pathways. Deubiquitinating enzymes (DUBs) have recently been attracting greater attention because these proteins are interesting in their function, which is involved in many cellular pathways [2].
Human Ub is a small (8 kDa) and abundant protein consisting of 76 amino acids and comprising as much as 5% of total protein within a cell. This protein is highly conserved among eukaryotic organisms. Monoubiquitination, which is the connection of a single Ub molecule by its C-terminal glycine (G76) to the lysine residue of a substrate protein, influences protein localization, DNA repair, endocytosis, virus budding, and protein–protein interaction [3]. The Ub molecule contains seven lysine residues—K6, K11, K27, K29, K33, K48, and K63—and each of these can be used for the creation of poly-Ub chains by conjugation of Ub molecules in a process known as polyubiquitination. Another type, the M1-linked Ub chain, can be formed by linkage of G76 of the connecting Ub to the N-terminal methionine of a Ub already attached in place of one of the lysine residues. Accordingly, this differential type of Ub connection or creation of poly-Ub chains results in various functions of the modified proteins [4,5]. In general, the ubiquitination can have a degradative or non-degradative purpose. Poly-Ub chains with Ub molecules bound via K48 or K11 determine substrates for proteasomal degradation [6,7]. A Ub chain attached via K63 has a regulatory function in several cell processes as a non-degradative signal, including in DNA reparation, signalization, endocytosis, vesicle transportation, and progression of the cell cycle [3,8]. It has also been reported that K33-linked chains are implicated in intracellular trafficking [9], and, together with K6-linked Ub chains, they are involved in DNA repair [10,11]. Several studies report that K6-linked Ub chains are related to the autophagy process, mitophagy, and xenophagy [12,13]. K29-linked chains have been observed in a proteasome regulation role [14,15] and in other epigenetic regulation in connectivity with the deubiquitinase Trabid [16,17]. The linear M1 ubiquitination is an essential modification in nuclear factor κB (NF-κB) activation [18]. Although the ubiquitin system is being intensively studied, the functions of all types of ubiquitination are not fully understood. Partly, it is because the diverse attachment alternatives give rise to a huge number of a possible complex poly-Ub chain structure (Figure 1).
In addition to Ub itself, other important molecules play vital roles in the ubiquitination processes. These are ubiquitin-like (Ubl) molecules, such as small ubiquitin-related modifier (SUMO), bacterial protein ThiS, and neural precursor cell expressed, developmentally downregulated 8 (NEDD8). Similar to Ub, these are involved in various cellular processes, such as transcriptional regulation, DNA repair, apoptosis, or protein stability [19,20]. Pathogens often target these small molecules during infection because they are involved in critical signaling pathways within the host cell and because disruption of the ubiquitination process could be advantageous to the pathogens [21].
2.1. Ubiquitination Process
The ubiquitination process consists of a cascade of enzymatic reactions depending on three key enzymes: Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), and Ub-ligase (E3). As mentioned above, ubiquitination is a reversible process, thus similar to some other post-translational modifications, so DUBs are also essential counterparts to these enzymes in the whole process. The initial step involves ATP-dependent Ub activation consisting in acyl-adenylation on the Ub C-terminus. The following part of the process includes transferring Ub to the E2 cysteine active site. The third enzyme, Ub-ligase, is able to recognize the target protein and most typically forms an isopeptide bond between G76 of Ub and lysine of the target protein (Figure 2). The individual key enzymes involved in the ubiquitination system will be described briefly for better understanding of the ubiquitination process.
2.2. Enzymes Participating in Ubiquitin System
Many different enzymes are included in the ubiquitin system. Surprisingly, the enzymes and components of the ubiquitination system comprise about 5% of the human proteome [5]. These include two Ub-activating enzymes [23], more than 50 Ub-conjugating enzymes, and several hundred Ub-ligases. The enormous number of different ubiquitination enzymes and varying complexity of Ub chains enable the formation of a subtle and complex regulation system tunable on many regulation levels and consisting of different products [24].
2.3. Ubiquitin Ligases
Ub-ligases comprise the biggest group of enzymes involved in the ubiquitination process. Their variability enables their modifying a wide variety of substrates. The primary function of Ub-ligases is transferring Ub to the target protein substrate [4,25]. The Ub ligation enzymes are divided into three families according to the domain each contains: Really Interesting New Gene (RING), homologous with E6-associated protein C-terminus (HECT), and RING-Between-RING (RBR) [26]. RING-type ligases work as scaffolds between the E2 enzyme and the target protein substrate and thus allow ubiquitination of the protein [27]. HECT-type ligases are different from RING-type ligases in the forming of a covalent thioester bond with Ub before its transfer to the substrate [28,29]. RBR-type ligases have both the RING and HECT domains, and their hybrid mechanism of action has been described [30,31,32].
2.4. Deubiquitinating Enzymes
The other enzymes playing a vital role in the ubiquitination system are DUBs, which are able to remove Ub from target substrates. DUBs are involved in many cellular processes and pathways, such as gene expression, apoptosis, cell cycle, DNA repair, and cytokine signaling [33]. DUBs were discovered in the group of metalloproteases, and mainly in the group of cysteine proteases. More than 90 DUBs have been identified and described in the human genome [2]. Cysteine DUBs are categorized into six subclasses according to the character of their ubiquitin-protease domains: Ub-specific proteases (USPs); Ub C-terminal hydrolases (UCHs); Machado–Joseph disease protein domain proteases (MJDs); ovarian tumor proteases (OTUs) [2]; a group of enzymes containing motif for interaction with the Ub-containing novel DUB family, known as MINDY [34]; and the newest family, ZUFSP (zinc finger with UFM1 specific peptidase) [35]. DUBs that were found in the metalloprotease group contain a domain that is referred to as JAMM (JAB1/MPN/Mov34 metalloenzyme) [2].
DUBs have widespread mechanisms of action for releasing Ub from the substrate. The Ub molecule is encoded by four genes (UBC, UBB, UBA52, and UBA80), and it is expressed as a linear chain of multiple Ub molecules. Subsequently, it has to be cut by DUBs. DUBs are essential for cleavage of polyubiquitin chains and complete removal of Ub chains from ubiquitinated proteins. These mechanisms lead to the maintenance of ubiquitin homeostasis in the cell, reverse Ub signaling, and correction of Ub-protein conjugates [36].
In contrast to other proteases, DUBs are generated as active enzymes. The catalytic activity of DUB cysteine proteases depends on two or three crucial amino acid residues forming a dyad or triad. These essential amino acids are from the group consisting of cysteine, histidine, aspartate, and asparagine. A variety of structures with the catalytic activity of DUBs has been reported [36]. Regulation of DUB activity is mediated by post-translational modifications, allosteric interactions, and subcellular localization. The catalytic triad of some DUBs has to be converted to active conformation by binding to a substrate, otherwise it remains inactive [2,36].
DUBs are able to recognize many ubiquitin-like molecules, isopeptides, and linear peptides, as well as various types of Ub linkage and chain structures. All DUB catalytic domains have a primary Ub-binding domain that encompasses interaction with the distal Ub in a poly-Ub chain. Moreover, each lysine residue in Ub molecule has a unique sequence that can be recognized by DUBs [36].
3. Immune Signaling Regulated by Ubiquitination
Ubiquitination is one of the crucial mechanisms of inflammation regulation (Figure 3) [37]. The pattern-recognition receptors (PRRs) mediate the recognition of pathogens in the innate immune response. Among them are nucleotide oligomerization domain (NOD)-like receptors, toll-like receptors (TLRs), and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) [38]. All of these receptors are able to induce NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, which activate the expression of proinflammatory cytokines [39].
One of the major proinflammatory cytokines is tumor necrosis factor (TNF), which induces a strong response and stimulates the recruitment of immune cells to sites of infection or damaged tissue. TNF binds tumor necrosis factor receptor 1 (TNFR1) to trigger a cascade of signaling reactions leading to activation of the NF-κB and MAPK pathways, where Ub linkage is important for NF-κB functioning [40]. K11-, M1-, and K63-linked polyubiquitinations are promoted mainly on receptor-interacting protein 1 (RIP1) but also on TNFR-associated factor TRAF2. This process generates a binding platform for assembly of the distal signaling components, such as cellular inhibitors of apoptosis (c-IAP) [41,42,43]. TNF-stimulated signaling is negatively regulated by DUBs. A DUB known as A20 removes K63-linked poly-Ub chains from RIP1, whereas another DUB, Ub-thioesterase otulin, removes linear chains from RIP1 and NF-κB essential modifier (NEMO), the inhibitor of κB kinase (IKK) regulatory subunit [44,45]. These DUB activities restricting TNF-induced NF-κB and MAPK signaling result in gene expression of proinflammatory cytokines [39].
K63-linked polyubiquitination of signal proteins, such as IRAK-1 or TRAF6, is necessary for triggering immune response based on activation of NF-κB. Polyubiquitination of IRAK-1 and TRAF6 is induced by stimulation of interleukin-1 receptor (IL-1R) or of TLRs by pathogens [46]. If some pathogens are able to block or disrupt this ubiquitination, it gives them the advantage of escaping from the host’s immune response.
NOD-like receptors work as cytoplasmatic PRRs which are able to recognize cytoplasmatic bacterial products. These receptors detect bacterial peptidoglycans. After activation, NODs oligomerize and recruit the kinase RIP2 and CARD9 leading to activation of NF-κB and MAPK signalization by ubiquitination of NEMO [47]. Similar to previous signalizations, the deubiquitinases are involved in the regulation of NOD2-RIP2 activation, such as A20 and otulin [44,48]. NOD2 may play a role in activation of inflammasomes [49]. Mutation in NOD2 can cause autoimmune and inflammatory diseases [47].
Signaling associated with ubiquitination is also observed during overgrowth of microbes that are a common part of the body, and their excessive amount or their metabolite products can influence immune function or dysfunction. Functioning of enzymes associated with ubiquitination is important for self-tolerance in autoimmunity [40]. Ubiquitination is a critical step for the activation of much inflammatory signaling. For example, ubiquitin-induced excessive activation of NF-κB could lead to inflammatory bowel diseases, such as ulcerative colitis or Crohn’s disease, where pathogenic or commensal bacteria in the gastrointestinal tract can be the cause of immune dysregulation [50]. NF-κB is important for intestinal homeostasis and it is regulated by different ubiquitin ligases and deubiquitinating enzymes. In this sense, the ubiquitin-proteasome system is also important, as it is involved in ensuring the homeostasis of post-translational modifications [51]. Moreover, commensal bacterium Bacteroides fragilis is able to produce its own eukaryotic-like ubiquitin protein, which is used as bacterial toxin for killing other intestinal bacteria [21,52].
4. Host Ubiquitination Machinery as a Target for Pathogens
Intracellular pathogens have developed many molecular mechanisms that are helpful in their strategies, such as their abilities to proliferate intracellularly and to survive host immune responses. Pathogens are able to produce effector proteins affecting the host cell processes in favor of pathogen survival. These proteins, often referred to as virulence factors, either interfere with important host cell structures, such as actin, microtubule, and intermediate filament cytoskeletons, or they are involved in manipulating endocytic, secretory, or signaling pathways in host metabolism [53,54].
Possible mechanisms have been described of bacterial intervention into the host ubiquitin system through the enzyme cascade involving E1, E2, and especially E3 enzymes [55]. Nonetheless, the pathogen interaction in the Ub deconjugation process is not fully understood. In any event, DUBs are enzymes specialized in the cleavage of Ub molecules, and in most cases they do not cleave Ub-like molecules (SUMO or NEDD8). On the other hand, CE clan proteases serve as specific hydrolases of Ub or Ub-like modifications in eukaryotic cells [56]. CE clan proteases are also synthesized by bacteria and work as effector proteins with deubiquitinase, deSUMOylase, or acetyltransferase activity. These effectors have mainly K63-linkage-specific deubiquitinase activity, and they have been found in human pathogens, such as Salmonella, Escherichia, and Legionella [54]. Several known effector proteins produced by bacterial pathogens are described in Table 1.
4.1. Bacterial Influence on Host Ubiquitinating and Deubiquitinating Processes
The ability of Yersinia enterocolitica and Y. pseudotuberculosis to invade host cells depends on the expression and catalytic activity of the human DUB otubain 1 (OTUB1). The main function of OTUB1 is to stabilize the active form of a small GTPase RhoA that is involved in formation of stress fibers during the bacterial attack. Yersinia spp. produces multifunctional protein kinase A (YpkA), which interferes with several parts of host cell processes and is regarded as a virulence factor [82]. YpkA is able to interact with post-translationally modified OTUB1 and modulates its influence on RhoA. This effect leads to cytoskeletal rearrangements that could be involved in bacterial uptake [57].
The relationship of the Ub system to bacterial invasion is supported by a recent study that proved the importance of human Ub C-terminal hydrolase UCH-L1 in the entry of Salmonella enterica and Listeria monocytogenes to the host cell. UCH-L1 is involved in spontaneous formation of actin stress fibers, which facilitate the capture of bacteria and subsequent entry into the cells [83].
The DUB UCH-L5 level is increased in macrophages during S. enterica serovar Typhimurium infection, and this leads to greater inflammasome activity. S. Typhimurium upregulates the expression of UCH-L5 by an unknown mechanism and causes increased catalytic activity of caspase-1, which limits the IL-1β level. The details of the overall mechanism need to be further investigated. Insufficient production of IL-1β can lead to cell death and the subsequent release of bacteria from macrophages during infection [84].
Pathogenesis of Helicobacter pylori is associated with gastritis, peptic ulcer, and gastric cancer. H. pylori can affect the host ubiquitin–proteasome system by intervening into the expression and activity of host DUB USP7. A lowering of USP7 decreases the amounts of TRAF6 and tumor suppressor p53, which partly explains the origin of carcinogenesis during chronic infection of H. pylori [85].
Francisella tularensis is an intracellular pathogen of humans and small mammals causing the zoonotic disease tularemia [86,87]. F. tularensis is able to subvert innate immune signaling pathways by influencing K63 polyubiquitination of TRAF3 and TRAF6 (Figure 4) [88]. This process is dependent on a functional type VI secretion system (T6SS). Francisella T6SS is encoded by genes located at Francisella pathogenicity island (FPI), and T6SS is very important for bacterial virulence. Many critical effector proteins are secreted by T6SS for support of intracellular growth [89], and the whole area of proteins secreted by F. tularensis that affect host ubiquitination machinery needs to be explored further. Li et al. [90] observed the function of host E3 ligase HECTD3 during bacterial infection of Francisella novicida, Mycobacterium bovis, and Listeria monocytogenes. The HECTD3 regulates TRAF3 K63 polyubiquitination in innate immune signaling response. Increased host defense against infection upon silencing of HECTD3 ligase has been determined [90]. Another host protein with DUB activity, USP22, is required for the proliferation of F. tularensis in the cytosol, and the ubiquitin ligase CDC27 is important for intervention into phagosomal maturation [91]. Guo et al. studied the host defense mechanism of inflammasome activation in F. novicida infection. They identified ubiquitin ligase HUWE1 as an AIM2-interacting protein. HUWE1 mediates K27 polyubiquitination of AIM2, and other inflammasomes proteins as NLRP4 and NLRC4, which promotes ASC (Apoptosis-associated Speck-like Protein) speck formation and cleaves caspase-1. Altogether, HUWE1 helps with eliminating bacterial burden, activation of caspase-1, and production of IL-1β [92]. Further, F. tularensis disturbs the adaptive immune response by inducing macrophage prostaglandin E2 (PGE2) production, which directly operates on CD4 T cells by restriction of interferon-γ production [93]. The other effect is in the downregulation of ubiquitin-dependent major histocompatibility complex (MHC) class II expression in macrophages [94]. Francisella-induced PGE2 drives the production of soluble macrophage factor FTMΦSN inducing ubiquitination of MHC class II by up-regulation of ubiquitin ligase MARCH1 in macrophages (Figure 4) [95].
Bacterial deubiquitination enzymes or ubiquitin-like proteins may not always be direct effector proteins but may be an essential part of the mechanisms that serve to secrete other effector proteins and virulence factors. As in the case of Staphylococcus aureus, where ubiquitin-like protein EsaB has been detected, which is a part of the type VII secretion system and is essential for the secretion activity [96,97].
Oxidative stress is observed during many pathological conditions such as cancer, inflammation, and infection. The ubiquitin proteasome system regulates the nuclear erythroid 2-related factor NRF2 antioxidant response [98] that is part of the KEAP1-NRF2 pathway during the protective response to oxidative stress. The heme oxygenase-1 (HO-1) stabilizes NRF2 during oxidative stress, thus NRF2 is accumulated and the preferential genes for detoxification enzymes are translated [99]. HO-1 has beneficial effects for protection against oxidative injury, apoptosis regulation, or modulation of inflammation. HO-1 as a cytoprotective enzyme with an anti-inflammatory and antioxidant role is very interesting for bacterial and viral pathogens [100,101,102].
4.2. Bacterial Modulation of Ubiquitin-Mediated Autophagy during Infection
Autophagy is a lysosome-based degradation process, highly conserved from yeast to human, first termed by Christian de Duve at a conference in 1963 and later in his publication regarding lysosome function [103,104]. Autophagy is activated upon various cellular stresses, such as starvation, organelle damage, or the presence of intracellular bacteria. The main functions of autophagy consist in removing harmful substances (such as protein aggregates, damaged organelles, and intracellular pathogens). Dysfunction of the autophagy process has been associated with numerous diseases in humans, including infectious diseases, cancer, neurodegeneration, cardiovascular disorders, and aging [105,106,107]. The process of autophagy includes the initiation and formation of autophagosomes (double membrane vesicles), subsequent fusion of an autophagosome with the lysosome, and final autolysosome formation that is responsible for the degradation of target molecules [108,109]. Several types of autophagy have been explored, and macroautophagy is the type best described. Macroautophagy is regulated by autophagy-related (ATG) proteins, and other regulators are initiated by formation of the phagophore, where ATG proteins play a critical role. In addition to Ub-independent selective autophagy, where autophagy receptors directly bind to intracellular cargo, Ub-dependent selective autophagy has been described [110]. Target subjects are conjugated with Ub chains and undergo subsequent degradation. Depending upon the target macromolecules, there are different types of autophagy, which include cargos like protein aggregates (aggrephagy), damaged mitochondria (mitophagy), lysosome (lysophagy), and bacteria (xenophagy) [111].
Not surprisingly, Ub is involved in many processes of autophagy [110,112]. Aggregated proteins, bacteria, and damaged mitochondria are labeled with Ub chains and then are recognized by receptors on the autophagosomal membrane [113]. The initiation of autophagy is dependent on many key regulators, such as ULK1 complex, containing ULK, Atg13, FIP200, and Atg101 proteins. The maturation of autophagosome is dependent on Ub-like conjugation systems, such as microtubule-associated protein 1 light chain 3 (LC3) and the Atg12 systems [111,114]. The ubiquitination of cytosolic bacteria in mammalian cells seems to be necessary for recognition and subsequent degradation by the antimicrobial autophagy process. This interaction is mediated through the LC3-interacting region (LIR) and Ub; subsequently, phagophore and autophagosome are formed, and bacteria are degraded in autolysosome (Figure 5) [110,113].
Many bacteria are able to manipulate ubiquitination during autophagy or even prevent maturation of autophagosome and thus escape from autophagy-dependent degradation. Similar to other intracellular bacteria, L. monocytogenes can escape from the phagosome in this way, and it then replicates in the host cell’s cytosol. This step is achieved by quickly expressing virulence factors upon entry to the host cell [115]. L. monocytogenes synthetizes ActA protein on its surface, which acts as a shield against autophagy. A mutant strain lacking functioning ActA protein is meanwhile labeled by Ub and unable to escape autophagy-dependent degradation. ActA figures as one of the key virulence factors involved in intracellular motility and is implicated in autophagy evasion. ActA is capable of recruiting actin-related proteins complex, vasodilator-stimulated phosphoproteins, or actin to avoid autophagic recognition by bacterial ubiquitination [77]. Another virulence factor of L. monocytogenes, InlK, supports camouflaging the bacterium’s surface by binding host cytoplasmic major vault protein (MVP), thereby blocking ubiquitination of the bacteria such that xenophagy does not occur [78]. Together with another two phospholipases, PlcA and PlcB, L. monocytogenes can inhibit autophagosome maturation [116].
Shigella flexneri also inhibits the autophagy process. S. flexneri produces important proteins that play a role in autophagosomal escape during infection. S. flexneri is able to inhibit autophagy by way of VirG and IcsB proteins. During normal conditions, IcsB blocks the binding between bacterial VirG and the host Atg5 (part of a complex that acts as an E3 ligase, through which ubiquitin-like proteins are conjugated to the autophagosomal membrane), thereby leading to reduced efficacy of autophagy. When IcsB is inactivated in a mutant strain lacking IcsB, however, the interaction between VirG and Atg5 occurs, leading to autophagy’s activation [117]. A second role of IscB in inhibiting host immune response against Shigella infection by inhibiting a manner of LC3-associated phagocytosis was described later [72]. Additionally, another effector protein, VirA, inactivates Rab1 that is required for the early formation of phagosome [118].
The p62 protein was the first selected autophagy adaptor to be described in mammalian cells [119,120]. It is involved in proteasomal degradation of ubiquitinated proteins [121] and also is involved in autophagy of S. Typhimurium. Many substrates are generated by Salmonella during infection that are targets for ubiquitination and are recognized by p62, which leads to autophagy of bacteria. S. Typhimurium produces SseL, a deubiquitinating enzyme that ensures removal of Ub from bacterial protein products and prevents recruitment of the autophagy markers p62 and LC3 [62]. Other Salmonella effectors SseF and SseG are able to impair autophagy initiation via disrupting the Rab1 signaling pathway [122].
Legionella pneumophila eliminates host autophagy processes by the production of effector protein RavZ, which hydrolyzes a bond between Ub-like protein Atg8 and phosphatidylethanolamine (PE) [123]. Atg8 (LC3 in mammalian cells) is required for autophagosomal membrane formation, and its conjugation to PE is a critical step in autophagy [124]. Not only does RavZ contain a catalytic domain related to the Ub-like enzymes, it also possesses a phosphatidylinositol-3-phosphate (PI3P) binding domain through which it has the ability to interact with PI3P that is present on the autophagosome surface and is important for RavZ localization to the autophagosomes [125]. A later study proved that RavZ first extracts the LC3-PE complex from the membrane and then acts as a deconjugation enzyme [126]. RavZ deconjugation activity includes not only PE release from LC3-PE but also Ub hydrolysis from substrate [70].
An effector protein important in the bacteria’s evading Ub-mediated autophagy also has been described in the case of Streptococcus pyogenes. In contrast to other serotypes, the globally disseminated clone M1T1 of S. pyogenes group A is able to replicate in host cells. The M1T1 strain produces SpeB protein, a cysteine protease that is able to degrade the ubiquitin-LC3 adaptor proteins NDP52, p62, and NDR1 [71].
Induction of selective autophagy also occurs in the case of the human pathogen Staphylococcus aureus. S. aureus was considered to be an extracellular bacterium, but its ability to invade a host cell and replicate inside was observed [127]. S. aureus is subject to selective autophagy through labeling by ubiquitination and subsequently it is recognized by receptor proteins SQSTM1, OPTN and CALCOCO2 [128].
Francisella tularensis is recognized by autophagic machinery mediated by the ubiquitin-SQSTM1-LC3 pathway after phagosomal escape [129]. F. tularensis uses surface polysaccharide O-antigen as masking before autophagy recognition, whereupon bacteria can successfully survive and proliferate in the host cytosol (Figure 4) [130].
The Ub molecule is also essential for the virulence of Mycobacterium tuberculosis. As mentioned above, the host immune defense mechanisms use Ub for labeling pathogens to ensure Ub-mediated selective autophagy. Two of the surface proteins of M. tuberculosis that directly undergo ubiquitination are Rv1468c [131] and Rv0222 [132]. Deliberate ubiquitination of mycobacterial surfaces and autophagy induction may be a strategy to prevent excessive inflammatory response, allowing bacteria to survive long-term intracellularly [131]. M. tuberculosis secretes tyrosine phosphatase PtpA that binds host Ub leading to subsequent activation and works as a dephosphorylating enzyme targeting JNK and MAPK p38 resulting in suppressing innate immunity [133].
Many bacteria modulate host immune response through disruption of autophagy and thus successfully survive and multiplicate inside the host cell. De/ubiquitination, during autophagosome formation, is a critical process, and therefore all effector proteins secreted by bacteria and interfering with autophagy machinery can be potentially relevant for host–pathogen interaction. Below, we focus on effectors directly related to Ub processes.
4.3. Bacterial Effectors Intervene with Ubiquitination
4.3.1. The Effect of Yersinia on the Host Immune Response
Yersinia pestis is the causative agent of plague, while Y. enterocolitica and Y. pseudotuberculosis cause gastrointestinal diseases. Each of these utilizes the type three secretion system (T3SS) for translocation of virulence proteins into the host cells in order to subvert host response. These effector proteins are classified as Yersinia outer proteins (Yops) [134].
The first discovered effector protein with DUB activity was Yersinia outer protein J (YopJ) in Y. pseudotuberculosis [135]. Its dual functions consist of acetyltransferase and deubiquitinase activity. The acetyltransferase activity leads to inhibition of MAPK and NF-κB signaling pathways, which block the activation of proinflammatory response [58]. The deubiquitinating function of YopJ is used in negative regulation of the signaling pathway by removing Ub from TRAF2, TRAF6, and IκBα. YopJ has shown specificity to K63-linked poly-Ub chains that activate IKK, but it also cleaves K48-linked poly-Ub chains to inhibit proteasomal degradation of IκBα [59]. YopJ is an important virulence factor that is able to directly influence the immune response through deubiquitination activity in Yersinia spp. infection.
4.3.2. Salmonella Produces Various Effectors
The Salmonella genome encodes T3SS to establish itself inside the host cell and translocate numerous effectors to the host cell [136]. Upon entry to the host cell, Salmonella is enclosed in a vacuolar compartment termed the Salmonella-containing vacuole (SCV) [137].
HECT-like E3 ubiquitin ligase SopA is an effector protein with a regulatory role in host inflammatory response. SopA ligase activity seems to play a crucial role in the induction of enteritis, but the biochemical mechanism and its substrate remain unknown [60].
S. enterica serovar Typhimurium produces DUB enzymes SseL and AvrA, which influence virulence and survival of bacteria in host cells [138,139]. AvrA, a homolog of YopJ, has shown a similar function by means of acetyltransferase activity [140]. AvrA removes ubiquitin moieties from IκBα and β-catenin, which regulate the NF-κB and β-catenin signaling pathways. Stabilization of IκBα leads to inhibition of the signaling and thus inhibition of inflammatory response. The stabilized β-catenin increases transcriptional activity, activation of cell proliferation, and inhibition of cell apoptosis [63].
Cysteine protease SseL was found to be secreted through T3SS into the host cell and detected on the vacuolar membrane 6 h after infection. SseL has shown preferences for K63-linked Ub chains on both mono- and polyubiquitin substrates, thus suggesting its involvement in signaling pathways rather than protein degradation [138]. Pruneda et al. studied the cleavage specificity of recombinant SseL, finding that this enzyme is active mainly against K63-linked chains and less so against K48- and K11-linked chains [54]. SseL appears to be necessary for the modulation of host inflammatory response during infection. The deubiquitinase activity of this enzyme leads to suppression of IκBα ubiquitination, which prevents NF-κB activation [61]. A study by Geng et al. showed significantly increased virulence activity of SseL effector-expressing strains in inhibiting the NF-κB pathway during S. enterica serovar Pullorum infection in a chicken model [141]. Furthermore, SseL works together with SseJ effector protein and alters the localization of lipid transporter oxysterol binding protein 1 (OSBP1) in S. enterica serovar Typhimurium. Deletion of both effectors increases the release of bacteria into the cytoplasm, which proves their important influence on vacuolar stability [142].
4.3.3. Escherichia Effectors
Enterohemorrhagic E. coli produces E3 ubiquitin ligase effector NleL that is transported to the host cell through outer membrane vesicles (OMVs), where it can ubiquitinylate c-Jun N-terminal kinase (JNK) and thus could have a suppressing effect on the proinflammatory signaling pathway [64].
Intestinal pathogenic E. coli produces deubiquitinating enzyme ElaD, which is an ortholog of S. enterica serovar Typhimurium SseL effector. ElaD enzyme can interact as deubiquitinase with linear and unanchored K63-linked polyUb chains rather than K48- and K11-linked polyUb chains [54,76]. Thus, the enzymatic activity and biological role of ElaD might be similar to those of Salmonella SseL [143].
4.3.4. Miscellaneous Protein Effectors of Legionella
The intracellular pathogen L. pneumophila also produces effector proteins with deubiquitinating activities that are secreted into the host cell. This pathogen uses a type IV secretion system to translocate effectors into host cells [144].
L. pneumophila uses not only canonical but also noncanonical ubiquitination for its infectivity promotion. For this effect, L. pneumophila uses SidE family effectors, some of which work as Ub-ligase in a special type of ubiquitination [65,145]. The noncanonical type of ubiquitination connects Ub to serine residues of host substrates via a phosphoribosyl (PR) moiety. It has been shown that PR-ubiquitinated proteins are associated with endoplasmic reticulum remodeling [145]. Moreover, there were two Legionella’s effectors (DupA and DupB) acting as deubiquitinase for PR-linked Ub (DUPs) identified. These enzymes counteract the effector SidE with ligase activity and remove PR-Ub from substrates. This process was essential for recruiting fragmented endoplasmic reticulum to Legionella-containing vacuoles (LCVs) upon L. pneumophila infection [66].
A recent study revealed the structure and mechanism of SdeA-mediated Ub modification [146]. SdeA, a member of the SidE effector family, plays an essential role in bacterial intracellular replication.Its DUB activity was also observed. SdeA has more than one domain and is able to recognize both Ub and NEDD8 Ubl. The N-terminal domain is a key component in the deubiquitination of three different poly-Ub chains: K11, K48, and K63. SdeA also removes K63-linked chains from LCVs, whereby Legionella directly and negatively affects the process of autophagy [67]. Downstream domains catalyze the mentioned PR-ubiquitination of substrates [146,147].
Another Legionella effector protein is LotA deubiquitinase, a member of the OTU family. LotA consists of two distinctive cysteine protease domains. The first cleavage domain is specific toward K6-linked Ub dimers. The second cysteine catalytic domain mainly contributes to removing chains of K48- and K63-linked Ub. The main function of LotA is to remove Ub from LCVs, and thus it protects bacteria from host immune processes. Not surprisingly, this protein is important for Legionella’s optimal intracellular growth [68].
RavD is an L. pneumophila effector protein with specific hydrolase activity against linear Ub chains. RavD limits the accumulation of linear Ub chains on LCVs in host cells to inhibit the NF-κB pathway during infection [69]. Surprisingly, RavD also has a phosphatidylinositol-binding domain, which is used to suppress the endolysosomal maturation of LCVs [148].
4.3.5. Insidious Shigella Effectors
Shigella flexneri is a causative agent of bacterial dysentery, mainly occurring in developing countries. It is able to permeate the epithelial cell wall and subsequently to escape the host’s immune response. After being taken up by macrophages, it is able to induce their apoptosis and escape into the cytoplasm [149]. The bacterium produces an effector protein that can purposefully influence the E2-enzyme level in host ubiquitin machinery. Shigella OspG protein is secreted by the T3SS, which can delay the NF-κB signaling pathway, and thus it also decelerates the transcription of NF-κB genes [73].
Proteins from the IpaH family are E3 ligases secreted and translocated into the host cells [150]. The IpaH Ub ligases antagonize activity of the linear Ub chain assembly complex (LUBAC), which acts as a Ub-ligase complex. The LUBAC enzymes generate M1 poly-Ub chains that play a role in the antimicrobial and proinflammatory signaling pathways [151]. IpaH ligases inhibit NF-κB signaling [75,152,153]; they participate in the recruitment of autophagy receptors OPTN, NDP52, and p62; and this helps Shigella to escape from xenophagy [74,154].
4.3.6. Chlamydia DUB Effectors
Chlamydia trachomatis is an obligate intracellular bacterial pathogen that causes various human diseases. Chlamydiae are able to avoid alarming the host immune system. One mechanism that C. trachomatis uses involves the expression of two enzymes, ChlaDub1 and dedicated ChlaDub2, with deubiquitinating and deneddylating activities [155]. Pruneda et al. observed that ChlaDub1 has a dual function, both K63-deubiquitinase and K-acetyltransferase activities, and, together with ChlaDub2, it intervenes in fragmentation of the host Golgi apparatus [156]. Like other intracellular pathogens, C. trachomatis suppresses NF-κB signaling activation by inhibiting IκBα ubiquitination and degradation, the actor in this case being ChlaDub1 [79]. The ChlaDUB proteins prevent Chlamydia-containing vacuoles’ ubiquitination and stabilize the antiapoptotic protein MCL-1 [157]. Another bacterial DUB, ChlaOTU, is a member of the OTU family, but its enzymatic specificity toward Ub linkage remains unresolved [80,81].
5. Ubiquitination and DUBs in Therapy
Due to importance of ubiquitination and DUBs in the host immune response during infection, these processes could be potential targets in therapies and effective treatments. Therapeutic substances could be helpful for the modulation of inflammatory conditions, antimicrobial and antiviral responses, and so in anticancer therapy. The examples of successful therapy in clinical use are specific inhibitors of proteasomal activity Bortezomib [158,159] or Carfilzomib [160]. They are applied in treatment of multiple myeloma cancer. Furthermore, several potential inhibitors of ubiquitin proteasome system in anticancer therapy are in clinical trials, for example the Nedd8-E1 enzyme inhibitor MLN4924 [161,162] or ubiquitin activating enzyme 1 inhibitor TAK-243 [163]. Nevertheless, no DUB targets have been found for clinical therapeutic use and they are needed to be further elucidated. One of them could be a deubiquitinase inhibitor WP1130, which has anti-infective effect against L. monocytogenes or noroviruses [164] and is also used as a treatment of malignancies [165]. The ubiquitin-proteasome system is important for virus replication. Many viruses encode their own DUBs; for example, severe acute respiratory syndrome coronavirus (SARS-CoV) encodes papain-like proteases [166]. Proteasome and DUBs inhibitors could be also helpful in the case of SARS-CoV-2 [167], where they could play an important role in antiviral therapy.
6. Conclusions
Ubiquitin and enzymes associated with ubiquitination machinery constitute an important component of signaling pathways in the immune host response to pathogen infection. While, for decades, attention has been devoted to acetylation and phosphorylation, other important post-translational modifications, such as ubiquitination, are now coming into the foreground. Transmitting a signal across the cell is a very intricate and complex process, and all events are more or less interconnected with one another. Effector molecules produced by dangerous pathogens are at the forefront of host immune response modulation. Among these, we find proteins with enzymatic activities that interfere with the ubiquitination process. Through this mechanism, pathogens can be provided with better conditions for survival and their spread within the host organism. When discussing the importance of ubiquitination on the host side, we must also emphasize the ability to deubiquitinate host target proteins on the pathogen side. The pathogen strategy for surviving when interfering with the ubiquitination system may substantially differ for every such pathogen. In some cases, however, bacteria effector proteins function similarly as results from the type of effector enzymatic activity. For instance, SseL, ElaD, and ShiCE effectors produced by S. Typhimurium, E. coli, and S. flexneri, respectively, are all primarily targeted toward K63-linked polyUb and suppress the NF-κB pathway in the innate immune response.
The ability of pathogens to control host immune response to their benefit by means of their effector molecules needs to be further explored. A better understanding of these molecular processes and possible identification of new effectors could be a good source of information for developing new therapeutic agents and vaccines against infections.
Author Contributions
Conceptualization, V.V.; writing—original draft preparation, V.V.; writing—review and editing, P.S., P.R., K.H.; visualization, V.V.; supervision, P.R., K.H., J.S.; project administration, V.V., J.S.; funding acquisition, J.S., V.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Specific Research grant of Ministry of Education, Youth and Sports of Czech Republic (SV/FVZ201802) and the long-term organization development plan, Medical Aspects of Weapons of Mass Destruction of the Faculty of Military Health Sciences, University of Defence, Ministry of Defence of the Czech Republic (DZRO-ZHN-2017).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, Functions, Mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Haglund, K.; Dikic, I. Ubiquitylation and Cell Signaling. EMBO J. 2005, 24, 3353–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.-W.; Ryu, K.-Y. Cellular Ubiquitin Pool Dynamics and Homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the Polyubiquitin Proteolytic Signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.T.; D’Andrea, A.D. Regulation of DNA Repair by Ubiquitylation. Nat. Rev. Mol. Cell Biol. 2006, 7, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-C.; Lee, Y.-R.; Lin, S.-Y.; Chang, L.-Y.; Tan, Y.P.; Hung, C.-C.; Kuo, J.-C.; Liu, C.-H.; Lin, M.-Y.; Xu, M.; et al. K33-Linked Polyubiquitination of Coronin 7 by Cul3-KLHL20 Ubiquitin E3 Ligase Regulates Protein Trafficking. Mol. Cell 2014, 54, 586–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, A.E.H.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.R.; Solomon, E. BRCA1: BARD1 Induces the Formation of Conjugated Ubiquitin Structures, Dependent on K6 of Ubiquitin, in Cells during DNA Replication and Repair. Hum. Mol. Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The Ubiquitin Ligase Parkin Mediates Resistance to Intracellular Pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Besche, H.C.; Sha, Z.; Kukushkin, N.V.; Peth, A.; Hock, E.-M.; Kim, W.; Gygi, S.; Gutierrez, J.A.; Liao, H.; Dick, L.; et al. Autoubiquitination of the 26S Proteasome on Rpn13 Regulates Breakdown of Ubiquitin Conjugates. EMBO J. 2014, 33, 1159–1176. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Xie, X.; Xiao, Y.; Hu, H.; Zou, Q.; Cheng, X.; Sun, S.-C. Epigenetic Regulation of the Expression of Il12 and Il23 and Autoimmune Inflammation by the Deubiquitinase Trabid. Nat. Immunol. 2016, 17, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Hamada, F.; Schwarz-Romond, T.; Bienz, M. Trabid, a New Positive Regulator of Wnt-Induced Transcription with Preference for Binding and Cleaving K63-Linked Ubiquitin Chains. Genes Dev. 2008, 22, 528–542. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of Linear Polyubiquitylation of NEMO in NF-KappaB Activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.S. Protein Modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef] [Green Version]
- Ribet, D.; Cossart, P. Ubiquitin, SUMO, and NEDD8: Key Targets of Bacterial Pathogens. Trends Cell Biol. 2018, 28, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Panasenko, O.O. Identification of Ubiquitinated Proteins. Mater. Methods 2014, 4, 827. [Google Scholar] [CrossRef]
- Mulder, M.P.C.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.-P.; Chang, J.-G.; Merkx, R.; Bialas, J.; Groettrup, M.; Vertegaal, A.C.O.; et al. A Cascading Activity-Based Probe Sequentially Targets E1–E2–E3 Ubiquitin Enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Neutzner, M.; Neutzner, A. Enzymes of Ubiquitination and Deubiquitination. Essays Biochem. 2012, 52, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Rape, M. Building Ubiquitin Chains: E2 Enzymes at Work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin Enzymes in the Regulation of Immune Responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 425–460. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING Finger Families of E3 Ubiquitin Ligases at a Glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Kumar, S. Mammalian HECT Ubiquitin-Protein Ligases: Biological and Pathophysiological Aspects. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.J.; Sixma, T.K. “Ubiquitylation: Mechanism and Functions” Review Series: RBR E3-Ligases at Work. EMBO Rep. 2014, 15, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 Ubiquitin Ligases: New Structures, New Insights, New Questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef] [Green Version]
- Lechtenberg, B.C.; Rajput, A.; Sanishvili, R.; Dobaczewska, M.K.; Ware, C.F.; Mace, P.D.; Riedl, S.J. Structure of a HOIP/E2~ubiquitin Complex Reveals RBR E3 Ligase Mechanism and Regulation. Nature 2016, 529, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.D. DUBs at a Glance. J. Cell Sci. 2009, 122, 2325–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the Chains: Structure and Function of the Deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.-C. Ubiquitin Signaling in Immune Responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-Like Receptor and RIG-1-Like Receptor Signaling. Available online: https://nyaspubs.onlinelibrary.wiley.com/doi/abs/10.1196/annals.1443.020 (accessed on 23 August 2019).
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the Immune System. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. C-IAP1 and c-IAP2 Are Critical Mediators of Tumor Necrosis Factor α (TNFα)-Induced NF-ΚB Activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [Green Version]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. C-IAP1 and UbcH5 Promote K11-Linked Polyubiquitination of RIP1 in TNF Signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Maecker, H.; Zobel, K.; Kömüves, L.G.; Deshayes, K.; Vucic, D. Cellular Inhibitors of Apoptosis Are Global Regulators of NF-ΚB and MAPK Activation by Members of the TNF Family of Receptors. Sci. Signal. 2012, 5, ra22. [Google Scholar] [CrossRef]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN Antagonizes LUBAC Signaling by Specifically Hydrolyzing Met1-Linked Polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-Ubiquitination and Ubiquitin Ligase Domains of A20 Downregulate NF-ΚB Signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Conze, D.B.; Wu, C.-J.; Thomas, J.A.; Landstrom, A.; Ashwell, J.D. Lys63-Linked Polyubiquitination of IRAK-1 Is Required for Interleukin-1 Receptor- and Toll-like Receptor-Mediated NF-KappaB Activation. Mol. Cell. Biol. 2008, 28, 3538–3547. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD Proteins: Regulators of Inflammation in Health and Disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Hitotsumatsu, O.; Ahmad, R.-C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The Ubiquitin-Editing Enzyme A20 Restricts Nucleotide-Binding Oligomerization Domain Containing 2-Triggered Signals. Immunity 2008, 28, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.R.; Levy, O. 3—Innate Immunity. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: London, UK, 2019; pp. 39–53. ISBN 978-0-7020-6896-6. [Google Scholar]
- Xiao, Y.; Huang, Q.; Wu, Z.; Chen, W. Roles of Protein Ubiquitination in Inflammatory Bowel Disease. Immunobiology 2020, 225, 152026. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Jobin, C. Ubiquitin Protein Modification and Signal Transduction: Implications for Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2005, 11, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Chatzidaki-Livanis, M.; Coyne, M.J.; Roelofs, K.G.; Gentyala, R.R.; Caldwell, J.M.; Comstock, L.E. Gut Symbiont Bacteroides Fragilis Secretes a Eukaryotic-Like Ubiquitin Protein That Mediates Intraspecies Antagonism. mBio 2017, 8, e01902-17. [Google Scholar] [CrossRef] [Green Version]
- Rytkönen, A.; Holden, D.W. Bacterial Interference of Ubiquitination and Deubiquitination. Cell Host Microbe 2007, 1, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruneda, J.N.; Durkin, C.H.; Geurink, P.P.; Ovaa, H.; Santhanam, B.; Holden, D.W.; Komander, D. The Molecular Basis for Ubiquitin and Ubiquitin-like Specificities in Bacterial Effector Proteases. Mol. Cell 2016, 63, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, L.A.; Edelmann, M.J. Ubiquitination as an Efficient Molecular Strategy Employed in Salmonella Infection. Front. Immunol. 2014, 5, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronau, J.A.; Beckmann, J.F.; Hochstrasser, M. Substrate Specificity of the Ubiquitin and Ubl Proteases. Cell Res. 2016, 26, 441–456. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, M.J.; Kramer, H.B.; Altun, M.; Kessler, B.M. Post-Translational Modification of the Deubiquitinating Enzyme Otubain 1 Modulates Active RhoA Levels and Susceptibility to Yersinia Invasion. FEBS J. 2010, 277, 2515–2530. [Google Scholar] [CrossRef]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ Acetylates and Inhibits Kinase Activation by Blocking Phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Monack, D.M.; Kayagaki, N.; Wertz, I.; Yin, J.; Wolf, B.; Dixit, V.M. Yersinia Virulence Factor YopJ Acts as a Deubiquitinase to Inhibit NF-ΚB Activation. J. Exp. Med. 2005, 202, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Higashide, W.M.; McCormick, B.A.; Chen, J.; Zhou, D. The Inflammation-Associated Salmonella SopA Is a HECT-like E3 Ubiquitin Ligase. Mol. Microbiol. 2006, 62, 786–793. [Google Scholar] [CrossRef]
- Negrate, G.L.; Faustin, B.; Welsh, K.; Loeffler, M.; Krajewska, M.; Hasegawa, P.; Mukherjee, S.; Orth, K.; Krajewski, S.; Godzik, A.; et al. Salmonella Secreted Factor L Deubiquitinase of Salmonella Typhimurium Inhibits NF-ΚB, Suppresses IκBα Ubiquitination and Modulates Innate Immune Responses. J. Immunol. 2008, 180, 5045–5056. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, F.S.; Thomas, M.; Sachse, M.; Santos, A.J.M.; Figueira, R.; Holden, D.W. The Salmonella Deubiquitinase SseL Inhibits Selective Autophagy of Cytosolic Aggregates. PLoS Pathog. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Ye, Z.; Petrof, E.O.; Boone, D.; Claud, E.C.; Sun, J. Salmonella Effector AvrA Regulation of Colonic Epithelial Cell Inflammation by Deubiquitination. Am. J. Pathol. 2007, 171, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Sirisaengtaksin, N.; O’Donoghue, E.J.; Jabbari, S.; Roe, A.J.; Krachler, A.M. Bacterial Outer Membrane Vesicles Provide an Alternative Pathway for Trafficking of Type III Secreted Effectors into Epithelial Cells. bioRxiv 2018, 415794. [Google Scholar] [CrossRef]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e13. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Liu, Y.; Gonzalez, A.; Bonn, F.; Liu, Y.; Rogov, V.V.; Heinz, M.; Stolz, A.; Hummer, G.; et al. Regulation of Phosphoribosyl-Linked Serine Ubiquitination by Deubiquitinases DupA and DupB. Mol. Cell 2020, 77, 164–179.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedlo, M.J.; Qiu, J.; Tan, Y.; Paul, L.N.; Luo, Z.-Q.; Das, C. Structural Basis of Substrate Recognition by a Bacterial Deubiquitinase Important for Dynamics of Phagosome Ubiquitination. Proc. Natl. Acad. Sci. USA 2015, 112, 15090–15095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubori, T.; Kitao, T.; Ando, H.; Nagai, H. LotA, a Legionella Deubiquitinase, Has Dual Catalytic Activity and Contributes to Intracellular Growth. Cell. Microbiol. 2018, 20, e12840. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Wang, X.; Huang, C.; Xu, D.; Wang, Z.; Zhou, Y.; Zhu, Y. A Bacterial Effector Deubiquitinase Specifically Hydrolyses Linear Ubiquitin Chains to Inhibit Host Inflammatory Signalling. Nat. Microbiol. 2019, 4, 1282–1293. [Google Scholar] [CrossRef]
- Kubori, T.; Bui, X.T.; Hubber, A.; Nagai, H. Legionella RavZ Plays a Role in Preventing Ubiquitin Recruitment to Bacteria-Containing Vacuoles. Front. Cell. Infect. Microbiol. 2017, 7, 384. [Google Scholar] [CrossRef]
- Nakagawa, I. Streptococcus Pyogenes Escapes from Autophagy. Cell Host Microbe 2013, 14, 604–606. [Google Scholar] [CrossRef] [Green Version]
- Baxt, L.A.; Goldberg, M.B. Host and Bacterial Proteins That Repress Recruitment of LC3 to Shigella Early during Infection. PLoS ONE 2014, 9, e94653. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Lenzen, G.; Page, A.-L.; Legrain, P.; Sansonetti, P.J.; Parsot, C. The Shigella Flexneri Effector OspG Interferes with Innate Immune Responses by Targeting Ubiquitin-Conjugating Enzymes. Proc. Natl. Acad. Sci. USA 2005, 102, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Mostowy, S.; Sancho-Shimizu, V.; Hamon, M.A.; Simeone, R.; Brosch, R.; Johansen, T.; Cossart, P. P62 and NDP52 Proteins Target Intracytosolic Shigella and Listeria to Different Autophagy Pathways. J. Biol. Chem. 2011, 286, 26987–26995. [Google Scholar] [CrossRef] [Green Version]
- Okuda, J.; Toyotome, T.; Kataoka, N.; Ohno, M.; Abe, H.; Shimura, Y.; Seyedarabi, A.; Pickersgill, R.; Sasakawa, C. Shigella Effector IpaH9.8 Binds to a Splicing Factor U2AF(35) to Modulate Host Immune Responses. Biochem. Biophys. Res. Commun. 2005, 333, 531–539. [Google Scholar] [CrossRef]
- Catic, A.; Misaghi, S.; Korbel, G.A.; Ploegh, H.L. ElaD, a Deubiquitinating Protease Expressed by E. Coli. PLoS ONE 2007, 2, e381. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Ogawa, M.; Hain, T.; Yoshida, M.; Fukumatsu, M.; Kim, M.; Mimuro, H.; Nakagawa, I.; Yanagawa, T.; Ishii, T.; et al. Listeria Monocytogenes ActA-Mediated Escape from Autophagic Recognition. Nat. Cell Biol. 2009, 11, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Mostowy, S.; Louaka, A.S.; Gouin, E.; Nahori, M.-A.; Wiemer, E.A.C.; Dussurget, O.; Cossart, P. Recruitment of the Major Vault Protein by InlK: A Listeria Monocytogenes Strategy to Avoid Autophagy. PLoS Pathog. 2011, 7, e1002168. [Google Scholar] [CrossRef]
- Le Negrate, G.; Krieg, A.; Faustin, B.; Loeffler, M.; Godzik, A.; Krajewski, S.; Reed, J.C. ChlaDub1 of Chlamydia Trachomatis Suppresses NF-KappaB Activation and Inhibits IkappaBalpha Ubiquitination and Degradation. Cell. Microbiol. 2008, 10, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Furtado, A.R.; Essid, M.; Perrinet, S.; Balañá, M.E.; Yoder, N.; Dehoux, P.; Subtil, A. The Chlamydial OTU Domain-Containing Protein ChlaOTU Is an Early Type III Secretion Effector Targeting Ubiquitin and NDP52. Cell. Microbiol. 2013, 15, 2064–2079. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Aravind, L.; Koonin, E.V. A Novel Superfamily of Predicted Cysteine Proteases from Eukaryotes, Viruses and Chlamydia Pneumoniae. Trends Biochem. Sci. 2000, 25, 50–52. [Google Scholar] [CrossRef]
- Laskowski-Arce, M.A.; Orth, K. The Elusive Activity of the Yersinia Protein Kinase A Kinase Domain Is Revealed. Trends Microbiol. 2007, 15, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Bassères, E.; Coppotelli, G.; Pfirrmann, T.; Andersen, J.B.; Masucci, M.; Frisan, T. The Ubiquitin C-Terminal Hydrolase UCH-L1 Promotes Bacterial Invasion by Altering the Dynamics of the Actin Cytoskeleton. Cell. Microbiol. 2010, 12, 1622–1633. [Google Scholar] [CrossRef]
- Kummari, E.; Alugubelly, N.; Hsu, C.-Y.; Dong, B.; Nanduri, B.; Edelmann, M.J. Activity-Based Proteomic Profiling of Deubiquitinating Enzymes in Salmonella-Infected Macrophages Leads to Identification of Putative Function of UCH-L5 in Inflammasome Regulation. PLoS ONE 2015, 10, e0135531. [Google Scholar] [CrossRef]
- Coombs, N.; Sompallae, R.; Olbermann, P.; Gastaldello, S.; Göppel, D.; Masucci, M.G.; Josenhans, C. Helicobacter Pylori Affects the Cellular Deubiquitinase USP7 and Ubiquitin-Regulated Components TRAF6 and the Tumour Suppressor P53. Int. J. Med. Microbiol. 2011, 301, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Oyston, P.C.F.; Green, M.; Titball, R.W. Tularemia. Clin. Microbiol. Rev. 2002, 15, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechous, R.D.; McCarthy, T.R.; Zahrt, T.C. Working toward the Future: Insights into Francisella Tularensis Pathogenesis and Vaccine Development. Microbiol. Mol. Biol. Rev. 2009, 73, 684–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putzova, D.; Panda, S.; Härtlova, A.; Stulík, J.; Gekara, N.O. Subversion of Innate Immune Responses by Francisella Involves the Disruption of TRAF3 and TRAF6 Signalling Complexes. Cell. Microbiol. 2017, 19, e12769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshraghi, A.; Kim, J.; Walls, A.C.; Ledvina, H.E.; Miller, C.N.; Ramsey, K.M.; Whitney, J.C.; Radey, M.C.; Peterson, S.B.; Ruhland, B.R.; et al. Secreted Effectors Encoded within and Outside of the Francisella Pathogenicity Island Promote Intramacrophage Growth. Cell Host Microbe 2016, 20, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, Y.; Liang, H.; Xu, T.; Kong, Y.; Huang, M.; Xiao, J.; Chen, X.; Xia, H.; Wu, Y.; et al. HECTD3 Mediates TRAF3 Polyubiquitination and Type I Interferon Induction during Bacterial Infection. J. Clin. Investig. 2018, 128, 4148–4162. [Google Scholar] [CrossRef] [Green Version]
- Akimana, C.; Al-Khodor, S.; Abu Kwaik, Y. Host Factors Required for Modulation of Phagosome Biogenesis and Proliferation of Francisella Tularensis within the Cytosol. PLoS ONE 2010, 5, e11025. [Google Scholar] [CrossRef]
- Guo, Y.; Li, L.; Xu, T.; Guo, X.; Wang, C.; Li, Y.; Yang, Y.; Yang, D.; Sun, B.; Zhao, X.; et al. HUWE1 Mediates Inflammasome Activation and Promotes Host Defense against Bacterial Infection. J. Clin. Investig. 2020, 130, 6301–6316. [Google Scholar] [CrossRef] [PubMed]
- Woolard, M.D.; Wilson, J.E.; Hensley, L.L.; Jania, L.A.; Kawula, T.H.; Drake, J.R.; Frelinger, J.A. Francisella Tularensis-Infected Macrophages Release Prostaglandin E2 That Blocks T Cell Proliferation and Promotes a Th2-like Response. J. Immunol. 2007, 178, 2065–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.E.; Katkere, B.; Drake, J.R. Francisella Tularensis Induces Ubiquitin-Dependent Major Histocompatibility Complex Class II Degradation in Activated Macrophages. Infect. Immun. 2009, 77, 4953–4965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, D.; Wilson, J.E.; Weih, K.A.; Ishido, S.; Harton, J.A.; Roche, P.A.; Drake, J.R. Francisella Tularensis Elicits IL-10 via a PGE2-Inducible Factor, to Drive Macrophage MARCH1 Expression and Class II Down-Regulation. PLoS ONE 2012, 7, e37330. [Google Scholar] [CrossRef]
- Casabona, M.G.; Buchanan, G.; Zoltner, M.; Harkins, C.P.; Holden, M.T.G.; Palmer, T. Functional Analysis of the EsaB Component of the Staphylococcus aureus Type VII Secretion System. Microbiology 2017, 163, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Warne, B.; Harkins, C.P.; Harris, S.R.; Vatsiou, A.; Stanley-Wall, N.; Parkhill, J.; Peacock, S.J.; Palmer, T.; Holden, M.T.G. The Ess/Type VII Secretion System of Staphylococcus aureus Shows Unexpected Genetic Diversity. BMC Genomics 2016, 17, 222. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2–Keap1 Antioxidant Response by the Ubiquitin Proteasome System: An Insight into Cullin-Ring Ubiquitin Ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear Heme Oxygenase-1 (HO-1) Modulates Subcellular Distribution and Activation of Nrf2, Impacting Metabolic and Anti-Oxidant Defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Hall, S.; Perrella, M.A. Role of Heme Oxygenase-1 in Microbial Host Defense. Cell. Microbiol. 2009, 11, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharn, C.R.; Collins, A.C.; Nair, V.R.; Stamm, C.E.; Marciano, D.K.; Graviss, E.A.; Shiloh, M.U. Heme Oxygenase-1 Regulates Inflammation and Mycobacterial Survival in Human Macrophages during M. Tuberculosis Infection. J. Immunol. 2016, 196, 4641–4649. [Google Scholar] [CrossRef] [Green Version]
- Lee, C. Therapeutic Modulation of Virus-Induced Oxidative Stress via the Nrf2-Dependent Antioxidative Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 6208067. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy Revisited: A Conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and Human Diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Murrow, L.; Debnath, J. Autophagy as a Stress-Response and Quality-Control Mechanism: Implications for Cell Injury and Human Disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. Autophagy: From Phenomenology to Molecular Understanding in Less than a Decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A Brief History of Autophagy from Cell Biology to Physiology and Disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin Signaling and Autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate MTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Cossart, P. Listeria Monocytogenes: Towards a Complete Picture of Its Physiology and Pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of Autophagy Mediated by PlcA or ActA Is Required for Listeria Monocytogenes Growth in Macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of Intracellular Shigella from Autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Zhu, Y.; Lu, Q.; Hu, L.; Zheng, Y.; Shao, F. Structurally Distinct Bacterial TBC-like GAPs Link Arf GTPase to Rab1 Inactivation to Counteract Host Defenses. Cell 2012, 150, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. P62 Links the Autophagy Pathway and the Ubiqutin–Proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.-Z.; Jiang, A.-J.; Mao, A.-W.; Feng, Y.; Wang, W.; Li, J.; Zhang, X.; Xing, K.; Peng, X. The Salmonella Effectors SseF and SseG Inhibit Rab1A-Mediated Autophagy to Facilitate Intracellular Bacterial Survival and Replication. J. Biol. Chem. 2018, 293, 9662–9673. [Google Scholar] [CrossRef] [Green Version]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella Effector RavZ Inhibits Host Autophagy through Irreversible Atg8 Deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Horenkamp, F.A.; Kauffman, K.J.; Kohler, L.J.; Sherwood, R.K.; Krueger, K.P.; Shteyn, V.; Roy, C.R.; Melia, T.J.; Reinisch, K.M. The Legionella Anti-Autophagy Effector RavZ Targets the Autophagosome via PI3P- and Curvature-Sensing Motifs. Dev. Cell 2015, 34, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Pantoom, S.; Wu, Y.-W. Elucidation of the Anti-Autophagy Mechanism of the Legionella Effector RavZ Using Semisynthetic LC3 Proteins. eLife 2017, 6, e23905. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Fraunholz, M. Staphylococcus aureus Host Cell Invasion and Post-Invasion Events. Int. J. Med. Microbiol. IJMM 2010, 300, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Neumann, Y.; Bruns, S.A.; Rohde, M.; Prajsnar, T.K.; Foster, S.J.; Schmitz, I. Intracellular Staphylococcus aureus Eludes Selective Autophagy by Activating a Host Cell Kinase. Autophagy 2016, 12, 2069–2084. [Google Scholar] [CrossRef] [Green Version]
- Chong, A.; Wehrly, T.D.; Child, R.; Hansen, B.; Hwang, S.; Virgin, H.W.; Celli, J. Cytosolic Clearance of Replication-Deficient Mutants Reveals Francisella Tularensis Interactions with the Autophagic Pathway. Autophagy 2012, 8, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Case, E.D.R.; Chong, A.; Wehrly, T.D.; Hansen, B.; Child, R.; Hwang, S.; Virgin, H.W.; Celli, J. The Francisella O-Antigen Mediates Survival in the Macrophage Cytosol via Autophagy Avoidance. Cell. Microbiol. 2014, 16, 862–877. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium Tuberculosis Surface Protein Recruits Ubiquitin to Trigger Host Xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, J.; Li, J.; Yang, H.; Tang, T.; Liang, H.; Zuo, M.; Wang, J.; Liu, H.; Liu, F.; et al. Host-Mediated Ubiquitination of a Mycobacterial Protein Suppresses Immunity. Nature 2020, 577, 682–688. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.-X.; Ge, P.-P.; Li, J.; Wang, Q.; Gao, G.F.; Qiu, X.-B.; Liu, C.H. Mycobacterium Tuberculosis Suppresses Innate Immunity by Coopting the Host Ubiquitin System. Nat. Immunol. 2015, 16, 237–245. [Google Scholar] [CrossRef]
- Cornelis, G.R. The Yersinia Deadly Kiss. J. Bacteriol. 1998, 180, 5495–5504. [Google Scholar] [CrossRef] [Green Version]
- Orth, K. Function of the Yersinia Effector YopJ. Curr. Opin. Microbiol. 2002, 5, 38–43. [Google Scholar] [CrossRef]
- Lostroh, C.P.; Lee, C.A. The Salmonella Pathogenicity Island-1 Type III Secretion System. Microbes Infect. 2001, 3, 1281–1291. [Google Scholar] [CrossRef]
- Oh, Y.K.; Alpuche-Aranda, C.; Berthiaume, E.; Jinks, T.; Miller, S.I.; Swanson, J.A. Rapid and Complete Fusion of Macrophage Lysosomes with Phagosomes Containing Salmonella Typhimurium. Infect. Immun. 1996, 64, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Rytkönen, A.; Poh, J.; Garmendia, J.; Boyle, C.; Thompson, A.; Liu, M.; Freemont, P.; Hinton, J.C.D.; Holden, D.W. SseL, a Salmonella Deubiquitinase Required for Macrophage Killing and Virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 3502–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, A.P.; Petrof, E.O.; Kuppireddi, S.; Zhao, Y.; Xia, Y.; Claud, E.C.; Sun, J. Salmonella Type III Effector AvrA Stabilizes Cell Tight Junctions to Inhibit Inflammation in Intestinal Epithelial Cells. PLoS ONE 2008, 3, e2369. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 2008, 3, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Wang, Y.; Xue, Y.; Wang, H.; Cai, Y.; Zhang, J.; Barrow, P.; Pan, Z.; Jiao, X. The SseL Protein Inhibits the Intracellular NF-ΚB Pathway to Enhance the Virulence of Salmonella Pullorum in a Chicken Model. Microb. Pathog. 2019, 129, 1–6. [Google Scholar] [CrossRef]
- Kolodziejek, A.M.; Altura, M.A.; Fan, J.; Petersen, E.M.; Cook, M.; Brzovic, P.S.; Miller, S.I. Salmonella Translocated Effectors Recruit OSBP1 to the Phagosome to Promote Vacuolar Membrane Integrity. Cell Rep. 2019, 27, 2147–2156.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanns, T.; Hofmann, K. Bacterial DUBs: Deubiquitination beyond the Seven Classes. Biochem. Soc. Trans. 2019, 47, 1857–1866. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.P.; Andrews, H.L.; Wong, S.K.; Isberg, R.R. Conjugative Transfer by the Virulence System of Legionella Pneumophila. Science 1998, 279, 873–876. [Google Scholar] [CrossRef]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.-Q. Ubiquitination Independent of E1 and E2 Enzymes by Bacterial Effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Mu, Y.; Xie, Y.; Zhang, Y.; Han, Y.; Zhou, Y.; Wang, W.; Liu, Z.; Wu, M.; Wang, H.; et al. Structural Basis of Ubiquitin Modification by the Legionella Effector SdeA. Nature 2018, 557, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Puvar, K.; Iyer, S.; Sheedlo, M.J.; Das, C. Chapter Fifteen—Purification and functional characterization of the DUB domain of SdeA. In Methods in Enzymology; Hochstrasser, M., Ed.; Ubiquitin and Ubiquitin-Like Protein Modifiers; Academic Press: Cambridge, MA, USA, 2019; Volume 618, pp. 343–355. [Google Scholar]
- Pike, C.M.; Boyer-Andersen, R.; Kinch, L.N.; Caplan, J.L.; Neunuebel, M.R. The Legionella Effector RavD Binds Phosphatidylinositol-3-Phosphate and Helps Suppress Endolysosomal Maturation of the Legionella-Containing Vacuole. J. Biol. Chem. 2019, 294, 6405–6415. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.N.; Hilbi, H. Molecular Pathogenesis of Shigella Spp.: Controlling Host Cell Signaling, Invasion, and Death by Type III Secretion. Clin. Microbiol. Rev. 2008, 21, 134–156. [Google Scholar] [CrossRef] [Green Version]
- Ashida, H.; Toyotome, T.; Nagai, T.; Sasakawa, C. Shigella Chromosomal IpaH Proteins Are Secreted via the Type III Secretion System and Act as Effectors. Mol. Microbiol. 2007, 63, 680–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, F.; Iwai, K. LUBAC, a Novel Ubiquitin Ligase for Linear Ubiquitination, Is Crucial for Inflammation and Immune Responses. Microbes Infect. 2012, 14, 563–572. [Google Scholar] [CrossRef]
- Ashida, H.; Kim, M.; Schmidt-Supprian, M.; Ma, A.; Ogawa, M.; Sasakawa, C. A Bacterial E3 Ubiquitin Ligase IpaH9.8 Targets NEMO/IKKgamma to Dampen the Host NF-KappaB-Mediated Inflammatory Response. Nat. Cell Biol. 2010, 12, 66–73. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella Flexneri Suppresses NF-KB Activation by Inhibiting Linear Ubiquitin Chain Ligation. Nat. Microbiol. 2016, 1, 16084. [Google Scholar] [CrossRef] [Green Version]
- Noad, J.; von der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-Synthesized Linear Ubiquitin Chains Restrict Cytosol-Invading Bacteria by Activating Autophagy and NF-ΚB. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [Green Version]
- Misaghi, S.; Balsara, Z.R.; Catic, A.; Spooner, E.; Ploegh, H.L.; Starnbach, M.N. Chlamydia Trachomatis-Derived Deubiquitinating Enzymes in Mammalian Cells during Infection. Mol. Microbiol. 2006, 61, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Pruneda, J.N.; Bastidas, R.J.; Bertsoulaki, E.; Swatek, K.N.; Santhanam, B.; Clague, M.J.; Valdivia, R.H.; Urbé, S.; Komander, D. A Chlamydia Effector Combining Deubiquitination and Acetylation Activities Induces Golgi Fragmentation. Nat. Microbiol. 2018, 3, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Harrison, K.S.; Ramirez, Y.; Auer, D.; Chowdhury, S.R.; Prusty, B.K.; Sauer, F.; Dimond, Z.; Kisker, C.; Hefty, P.S.; et al. Chlamydia Trachomatis-Containing Vacuole Serves as Deubiquitination Platform to Stabilize Mcl-1 and to Interfere with Host Defense. eLife 2017, 6, e21465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (VelcadeTM) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Groen, K.; van de Donk, N.; Stege, C.; Zweegman, S.; Nijhof, I.S. Carfilzomib for Relapsed and Refractory Multiple Myeloma. Cancer Manag. Res. 2019, 11, 2663–2675. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. MLN4924: A Novel First-in-Class Inhibitor of NEDD8-Activating Enzyme for Cancer Therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef]
- Millennium Pharmaceuticals, Inc. MLN4924 for the Treatment of Acute Myelogenous Leukemia, Myelodysplastic Syndrome, and Acute Lymphoblastic Leukemia; Millennium Pharmaceuticals Inc.: Cambridge, MA, USA, 2013. [Google Scholar]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-Activating Enzyme Inhibition Induces an Unfolded Protein Response and Overcomes Drug Resistance in Myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef] [Green Version]
- Charbonneau, M.-E.; Gonzalez-Hernandez, M.J.; Showalter, H.D.; Donato, N.J.; Wobus, C.E.; O’Riordan, M.X.D. Small Molecule Deubiquitinase Inhibitors Promote Macrophage Anti-Infective Capacity. PLoS ONE 2014, 9, e104096. [Google Scholar] [CrossRef]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase Inhibition by Small-Molecule WP1130 Triggers Aggresome Formation and Tumor Cell Apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, H.A. Deubiquitination in Virus Infection. Virology 2007, 362, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The ubiquitination of substrates may vary in different constructions that play a role in various cell processes: protein interaction and localization (A), proteasomal degradation (B), NF-κB activation (C,D), DNA repair (C,E,F), lysosomal targeting (C), autophagy (E), intracellular trafficking (F) and epigenetic regulation (G), and many others still unknown functions (H). S—substrate, Ub—ubiquitin. Adapted from [5].
Figure 1.
The ubiquitination of substrates may vary in different constructions that play a role in various cell processes: protein interaction and localization (A), proteasomal degradation (B), NF-κB activation (C,D), DNA repair (C,E,F), lysosomal targeting (C), autophagy (E), intracellular trafficking (F) and epigenetic regulation (G), and many others still unknown functions (H). S—substrate, Ub—ubiquitin. Adapted from [5].

Figure 2.
Scheme of ubiquitin binding to the target protein [22]. Ub is activated by ATP-assisted acyl-adenylation, bound to the Ub-activating enzyme (E1), transferred to Ub-conjugating enzyme (E2), and attached to the target protein substrate (S) through Ub-ligase (E3). This cycle mechanism can be repeated for the creation of a polyubiquitin chain. ATP—adenosine triphosphate, AMP—adenosine monophosphate, PPi—pyrophosphate, Ub—ubiquitin. Adapted from [22].
Figure 2.
Scheme of ubiquitin binding to the target protein [22]. Ub is activated by ATP-assisted acyl-adenylation, bound to the Ub-activating enzyme (E1), transferred to Ub-conjugating enzyme (E2), and attached to the target protein substrate (S) through Ub-ligase (E3). This cycle mechanism can be repeated for the creation of a polyubiquitin chain. ATP—adenosine triphosphate, AMP—adenosine monophosphate, PPi—pyrophosphate, Ub—ubiquitin. Adapted from [22].

Figure 3.
Activation of innate immune signaling pathway by lipopolysaccharide (LPS) and tumor necrosis factor (TNF). LPS is bound by toll-like receptor 4 (TLR4), and then adaptor protein MyD88 interacts with IRAK protein kinases. Recruitment of IRAK complex results in association with another adaptor protein, TRAF6, and this leads to IκB kinase (IKK) activation. NF-κB is a functional transcription factor after IκBα phosphorylation and subsequent dissociation of the complex of NF-κB and IκBα. IκBα, an inhibitor of NF-κB, can also be degraded by way of ubiquitination. Ubiquitinated IκB is degraded in proteasome and NF-κB moves to the nucleus, where it activates the proinflammatory cytokine transcription. A different way of activating the NF-κB pathway shown here is through binding of TNF by TNF receptor 1 (TNFR1). Adaptor protein TRADD recruits receptor-binding protein 1 (RIP1) and TNFR-associated factor TRAF2. RIP1 activates IKK complex and thus also NF-κB pathway. It is shown that A20 and otulin interact with Ub chains in the signaling pathway.
Figure 3.
Activation of innate immune signaling pathway by lipopolysaccharide (LPS) and tumor necrosis factor (TNF). LPS is bound by toll-like receptor 4 (TLR4), and then adaptor protein MyD88 interacts with IRAK protein kinases. Recruitment of IRAK complex results in association with another adaptor protein, TRAF6, and this leads to IκB kinase (IKK) activation. NF-κB is a functional transcription factor after IκBα phosphorylation and subsequent dissociation of the complex of NF-κB and IκBα. IκBα, an inhibitor of NF-κB, can also be degraded by way of ubiquitination. Ubiquitinated IκB is degraded in proteasome and NF-κB moves to the nucleus, where it activates the proinflammatory cytokine transcription. A different way of activating the NF-κB pathway shown here is through binding of TNF by TNF receptor 1 (TNFR1). Adaptor protein TRADD recruits receptor-binding protein 1 (RIP1) and TNFR-associated factor TRAF2. RIP1 activates IKK complex and thus also NF-κB pathway. It is shown that A20 and otulin interact with Ub chains in the signaling pathway.

Figure 4.
Francisella tularensis manipulation of the host cell ubiquitination system. (A) F. tularensis requires host factor DUB USP22 for cytosol proliferation and E3 ligase CDC27 for intervention to phagosomal maturation. (B) Adaptive immune response is changed upon induction of host prostaglandine E2 (PGE2), where the E3 ligase MARCH1 regulates the ubiquitination of MHC class II. (C) Innate immune signalization is subverted by changes in the ubiquitination of TRAF3 and TRAF6 adaptors with E3 ligase HECTD3 regulating the K63 polyubiquitination of TRAF3. (D) F. tularensis surface O-antigen hinders the autophagy recognition that enables the bacteria proliferation in the cytosol of the host cell. (E) Host cell defense mechanism is positively influenced by inflammasome activation through E3 ligase HUWE1.
Figure 4.
Francisella tularensis manipulation of the host cell ubiquitination system. (A) F. tularensis requires host factor DUB USP22 for cytosol proliferation and E3 ligase CDC27 for intervention to phagosomal maturation. (B) Adaptive immune response is changed upon induction of host prostaglandine E2 (PGE2), where the E3 ligase MARCH1 regulates the ubiquitination of MHC class II. (C) Innate immune signalization is subverted by changes in the ubiquitination of TRAF3 and TRAF6 adaptors with E3 ligase HECTD3 regulating the K63 polyubiquitination of TRAF3. (D) F. tularensis surface O-antigen hinders the autophagy recognition that enables the bacteria proliferation in the cytosol of the host cell. (E) Host cell defense mechanism is positively influenced by inflammasome activation through E3 ligase HUWE1.

Figure 5.
A cargo for selective ubiquitin-dependent autophagy, e.g., bacteria, aggregated protein or damaged mitochondria, is labeled by Ub chain. The cargo is recognized by autophagy receptors on the phagosomal membrane and subsequently maturation leading to the emergence of autophagosome occurs. The autolysosome is formed after fusion of autophagosome with lysosome, where the cargo is degraded. LC3—microtubule-associated protein 1 light chain 3, Ub—ubiquitin.
Figure 5.
A cargo for selective ubiquitin-dependent autophagy, e.g., bacteria, aggregated protein or damaged mitochondria, is labeled by Ub chain. The cargo is recognized by autophagy receptors on the phagosomal membrane and subsequently maturation leading to the emergence of autophagosome occurs. The autolysosome is formed after fusion of autophagosome with lysosome, where the cargo is degraded. LC3—microtubule-associated protein 1 light chain 3, Ub—ubiquitin.


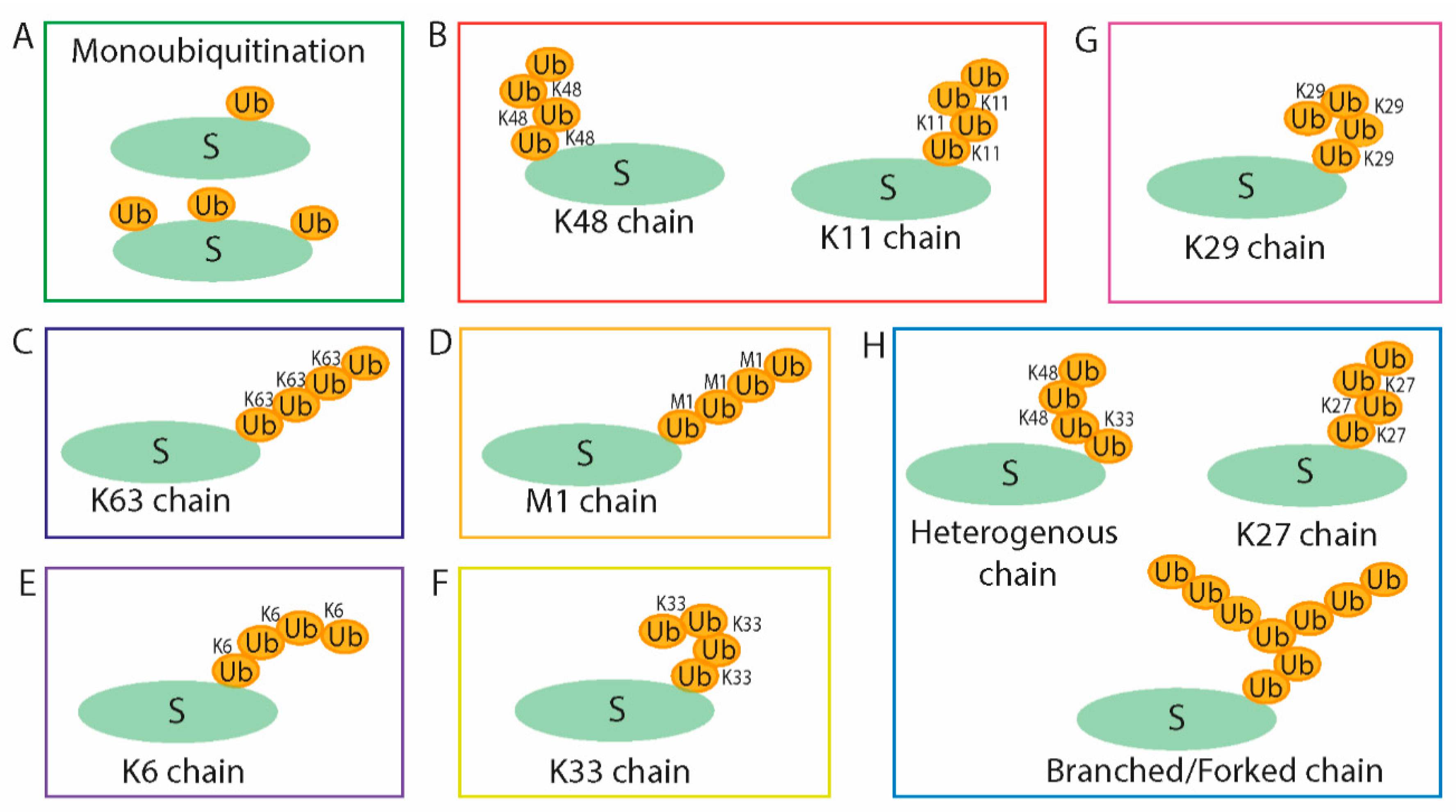{kind=link}
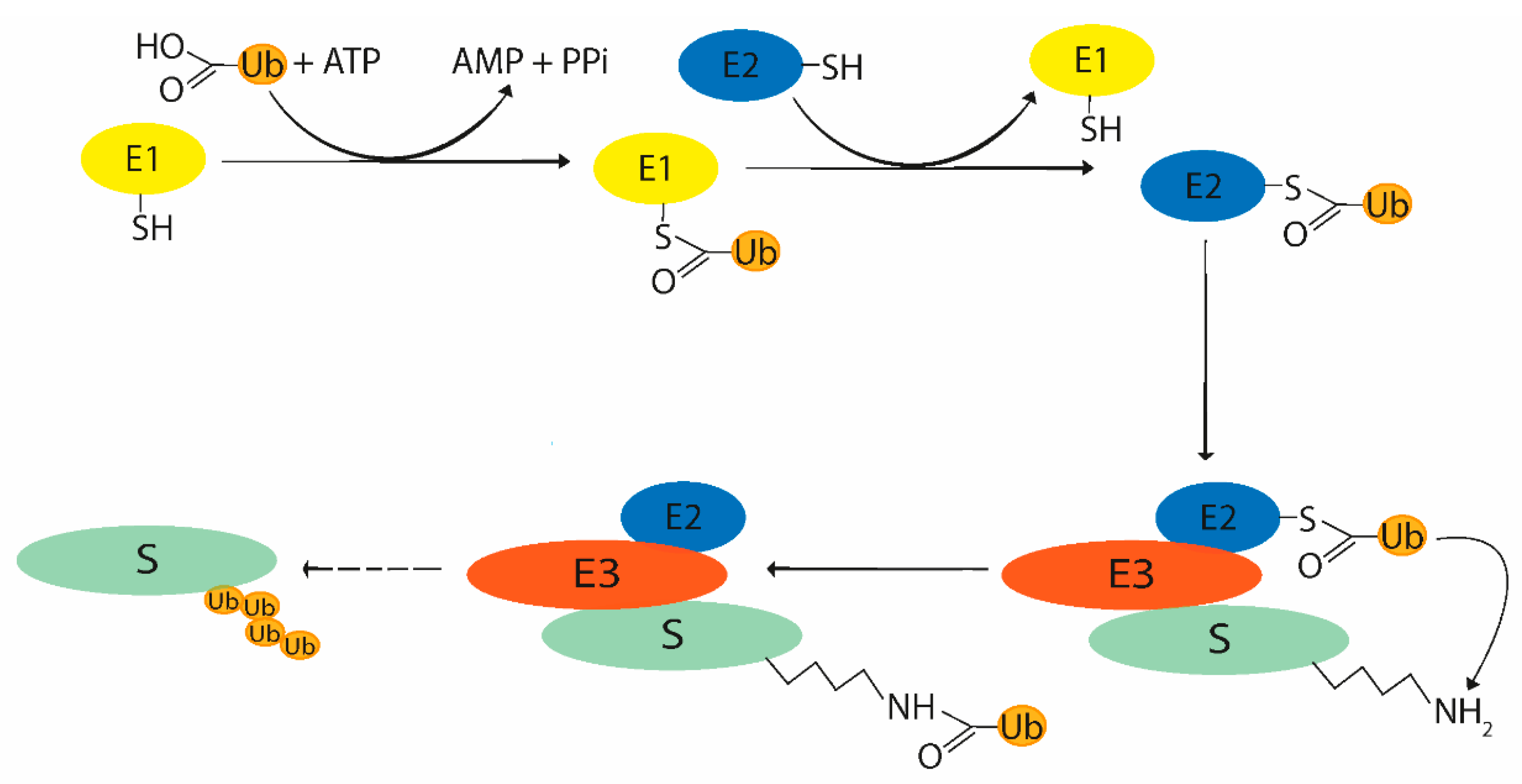{kind=link}
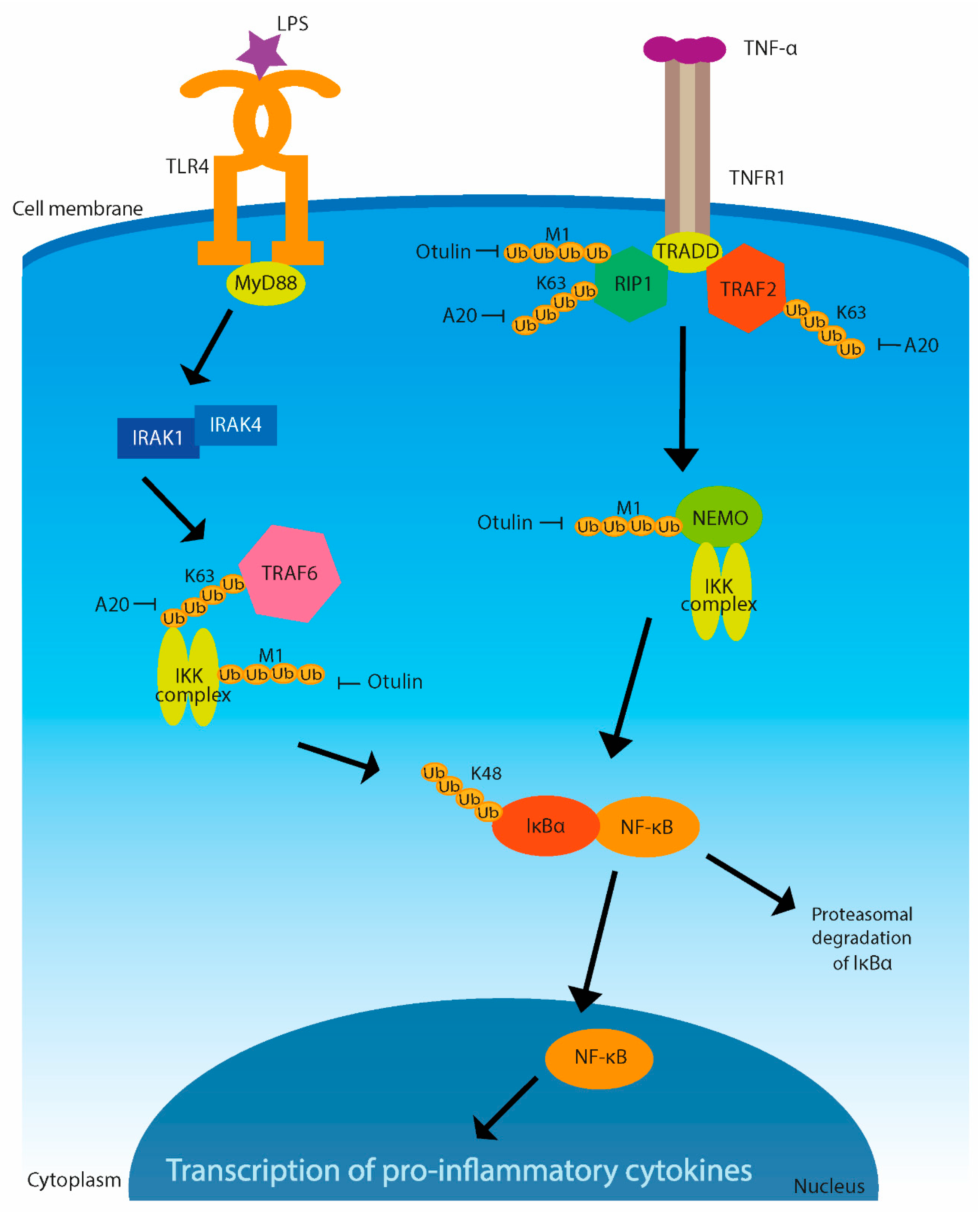{kind=link}
{kind=link}
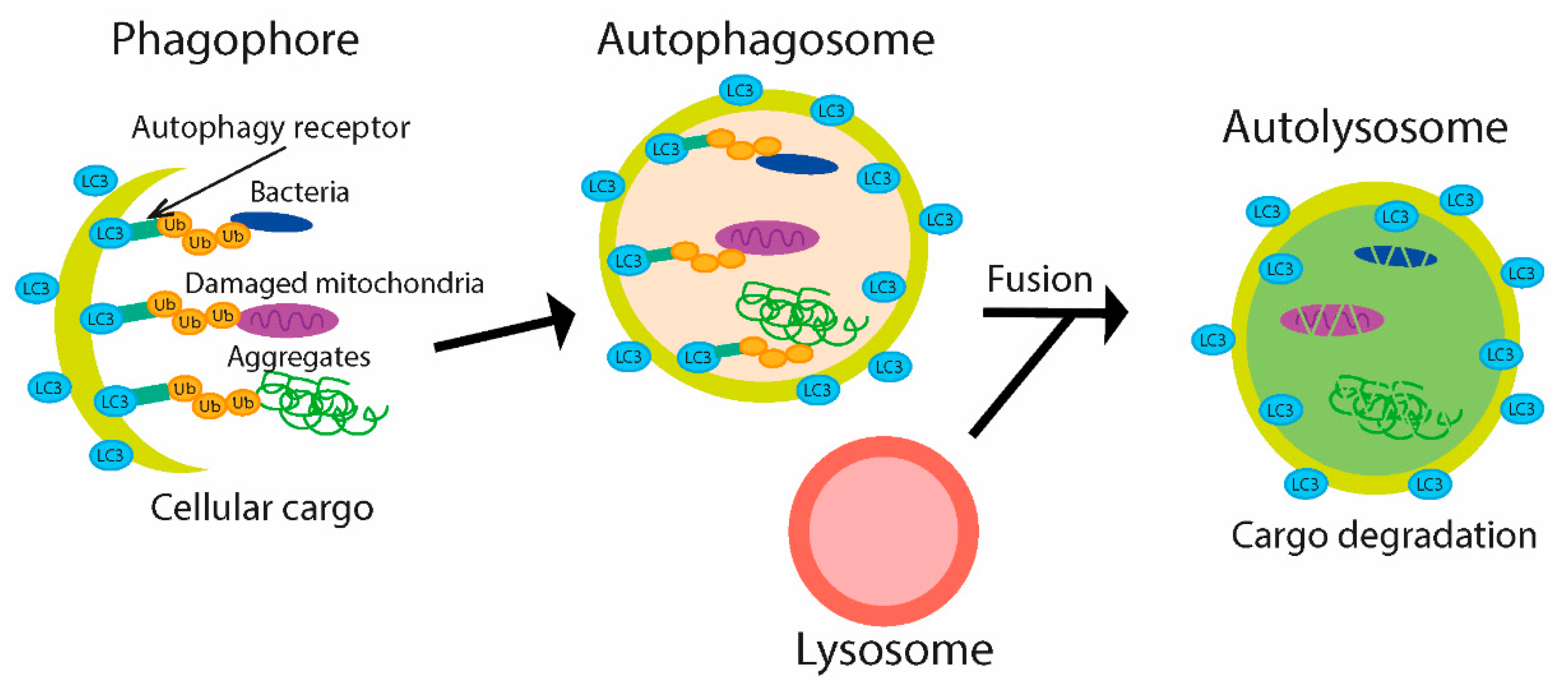{kind=link}
Table 1.
Bacterial effector proteins described in this review.
| Effector Protein | Bacteria | Role | Function | References |
|---|---|---|---|---|
| YpkA | Yersinia pseudotuberculosis, Y. enterocolitica | Protein kinase | Increasing of bacterial uptake | [57] |
| YopJ | Yersinia pseudotuberculosis | DUB, acetyltransferase | Suppression of MAPK and NF-κB pathway | [58,59] |
| SopA | Salmonella Typhimurium | E3 ligase | Unknown | [60] |
| SseL | Salmonella Typhimurium | DUB | Suppression of NF-κB pathway | [61] |
| Inhibition of autophagy | [62] | |||
| AvrA | Salmonella Typhimurium | DUB, acetyltransferase | Suppression of NF-κB pathway and stabilization of β-catenin | [63] |
| NleL | Escherichia coli | E3 ligase | Suppression of the inflammatory response | [64] |
| ElaD | Escherichia coli | DUB | Suppression of NF-κB pathway | [54] |
| SidE | Legionella pneumophila | E3 ligase | Regulation of phosphoribosyl ubiquitination, cytotoxicity | [65,66] |
| DupA, B | Legionella pneumophila | DUB | ||
| SdeA | Legionella pneumophila | DUB | Inhibition of autophagy | [67] |
| LotA | Legionella pneumophila | DUB | Suppression of phagosome maturation | [68] |
| RavD | Legionella pneumophila | DUB | Suppression of phagosome maturation and NF-κB pathway | [69] |
| RavZ | Legionella pneumophila | DUB | Inhibition of autophagy | [70] |
| SpeB | Streptococcus pyogenes | Cysteine protease | Inhibition of autophagy | [71] |
| IcsB | Shigella flexneri | Virulence factor | Inhibition of autophagy | [72] |
| OspG | Shigella flexneri | E2 enzyme | Suppression of NF-κB pathway | [73] |
| IpaH | Shigella flexneri | E3 ligase | Suppression of NF-κB pathway and autophagy | [74,75] |
| SchiCE | Shigella flexneri | DUB | Suppression of NF-κB pathway | [54,76] |
| ActA | Listeria monocytogenes | Virulence factor | Increasing of intracellular motility to avoiding autophagy | [77] |
| InlK | Listeria monocytogenes | Virulence factor | Inhibition of autophagy | [78] |
| ChlaDUB1 | Chlamydia trachomatis | DUB, acetyltransferase | Suppression of NF-κB pathway | [79] |
| ChlaDUB2 | Chlamydia trachomatis | DUB | Suppression of NF-κB pathway | |
| ChlaOTU | Chlamydia pneumonia | DUB | Unknown | [80,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vozandychova, V.; Stojkova, P.; Hercik, K.; Rehulka, P.; Stulik, J. The Ubiquitination System within Bacterial Host–Pathogen Interactions. Microorganisms 2021, 9, 638. https://doi.org/10.3390/microorganisms9030638
AMA Style
Vozandychova V, Stojkova P, Hercik K, Rehulka P, Stulik J. The Ubiquitination System within Bacterial Host–Pathogen Interactions. Microorganisms. 2021; 9(3):638. https://doi.org/10.3390/microorganisms9030638
Chicago/Turabian StyleVozandychova, Vera, Pavla Stojkova, Kamil Hercik, Pavel Rehulka, and Jiri Stulik. 2021. "The Ubiquitination System within Bacterial Host–Pathogen Interactions" Microorganisms 9, no. 3: 638. https://doi.org/10.3390/microorganisms9030638
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.