
Synthesis and Anticancer and Antiviral Activities of C-2′-Branched Arabinonucleosides
 , and
, and
Abstract
:1. Introduction
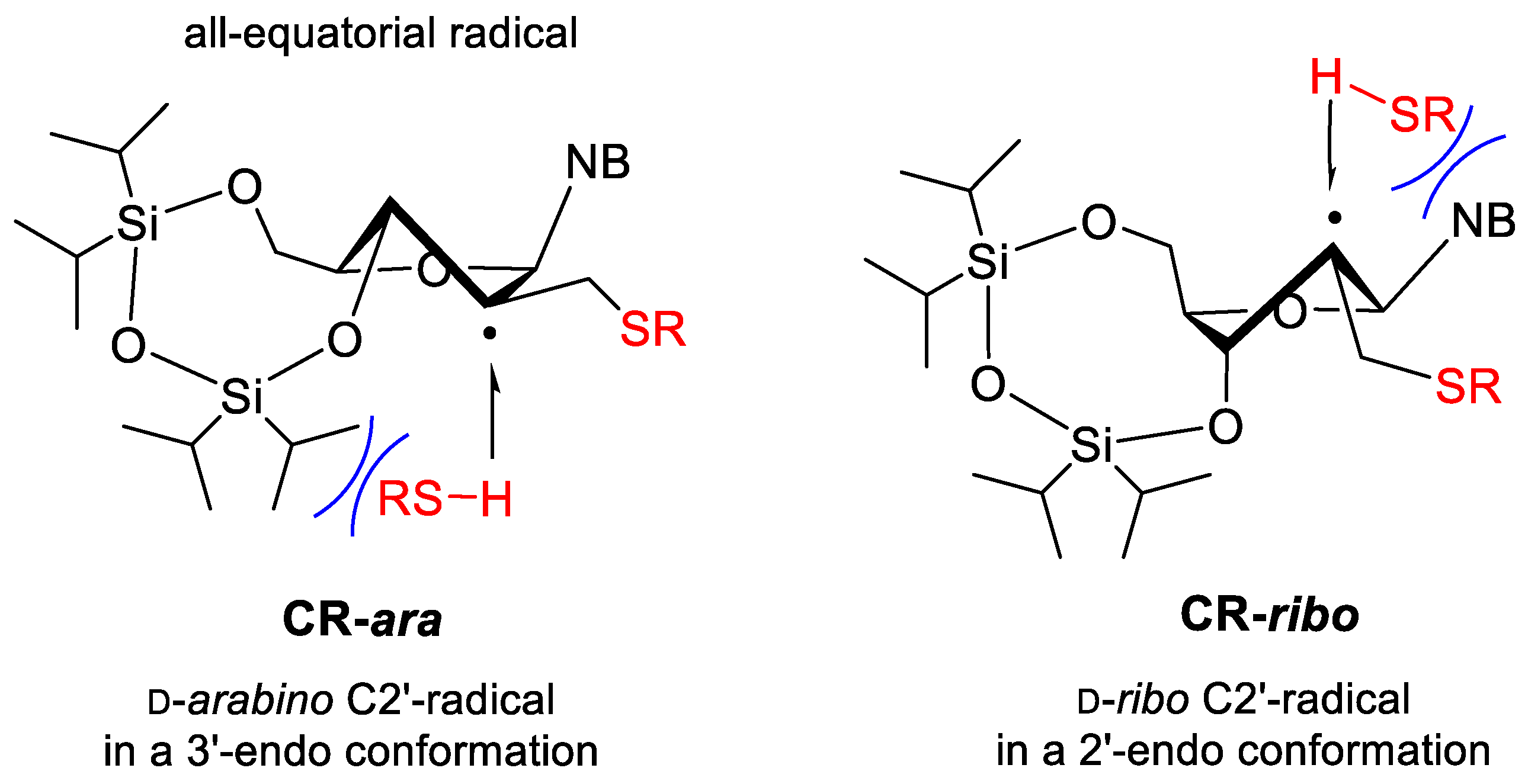
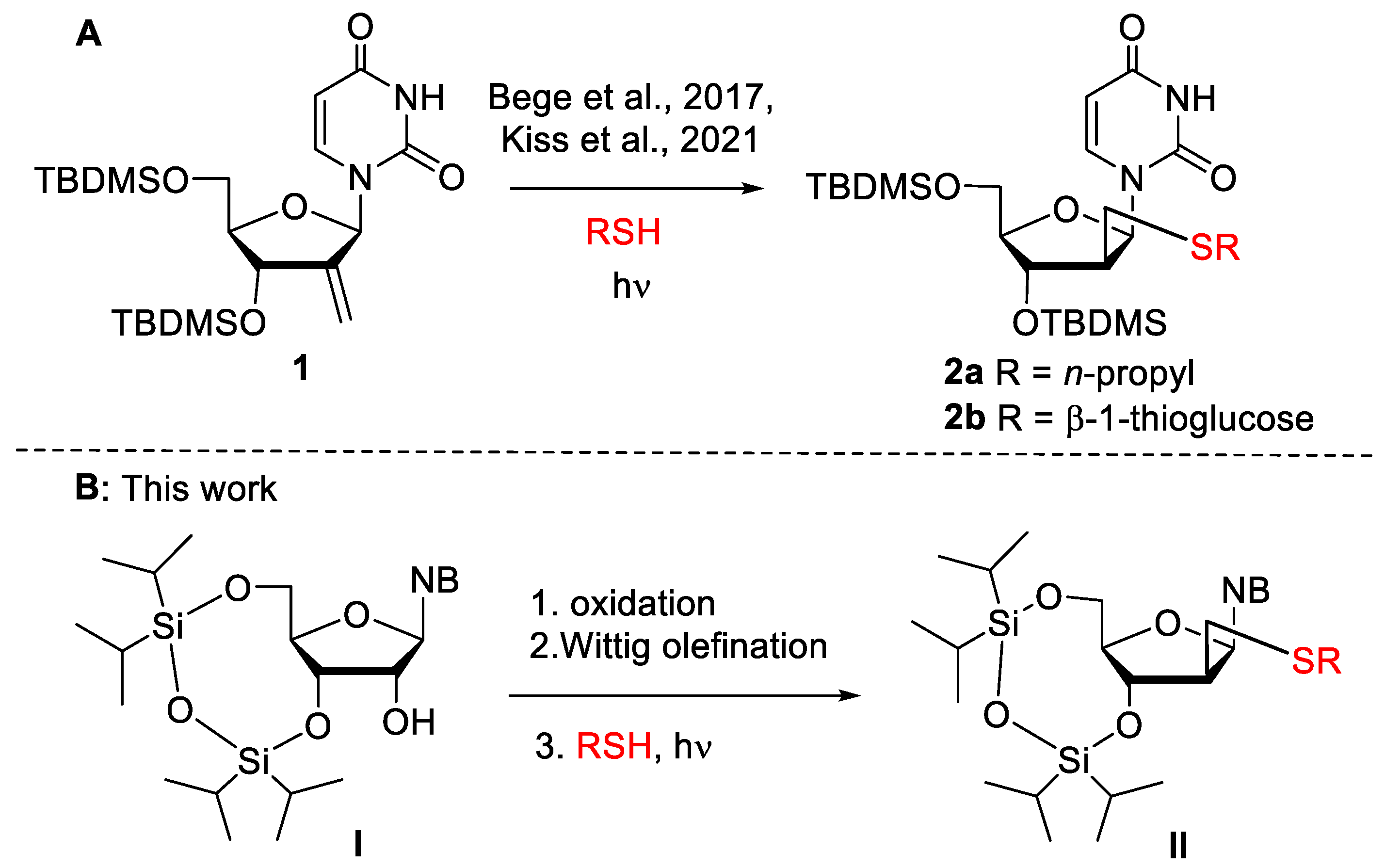
2. Results and Discussion
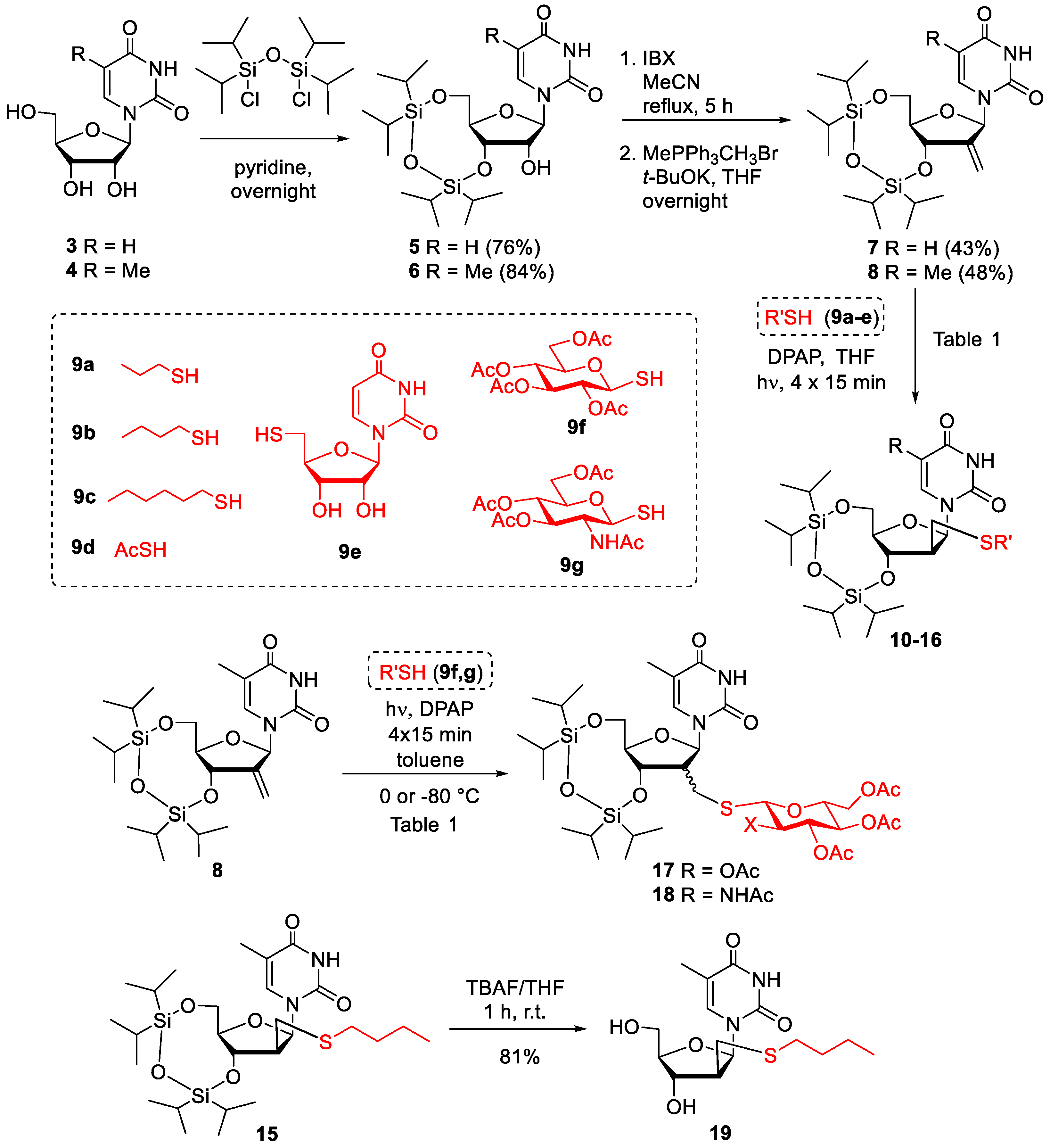
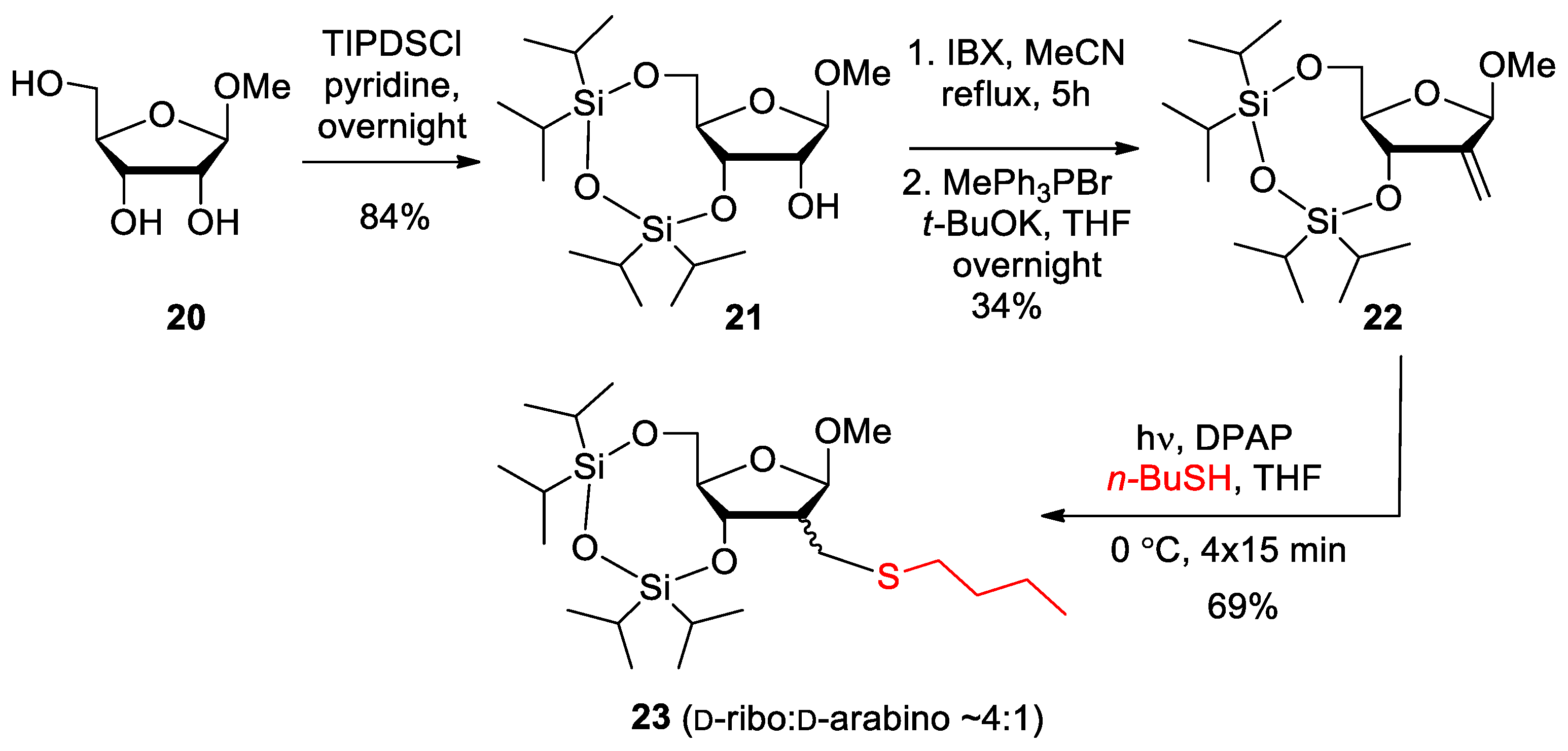
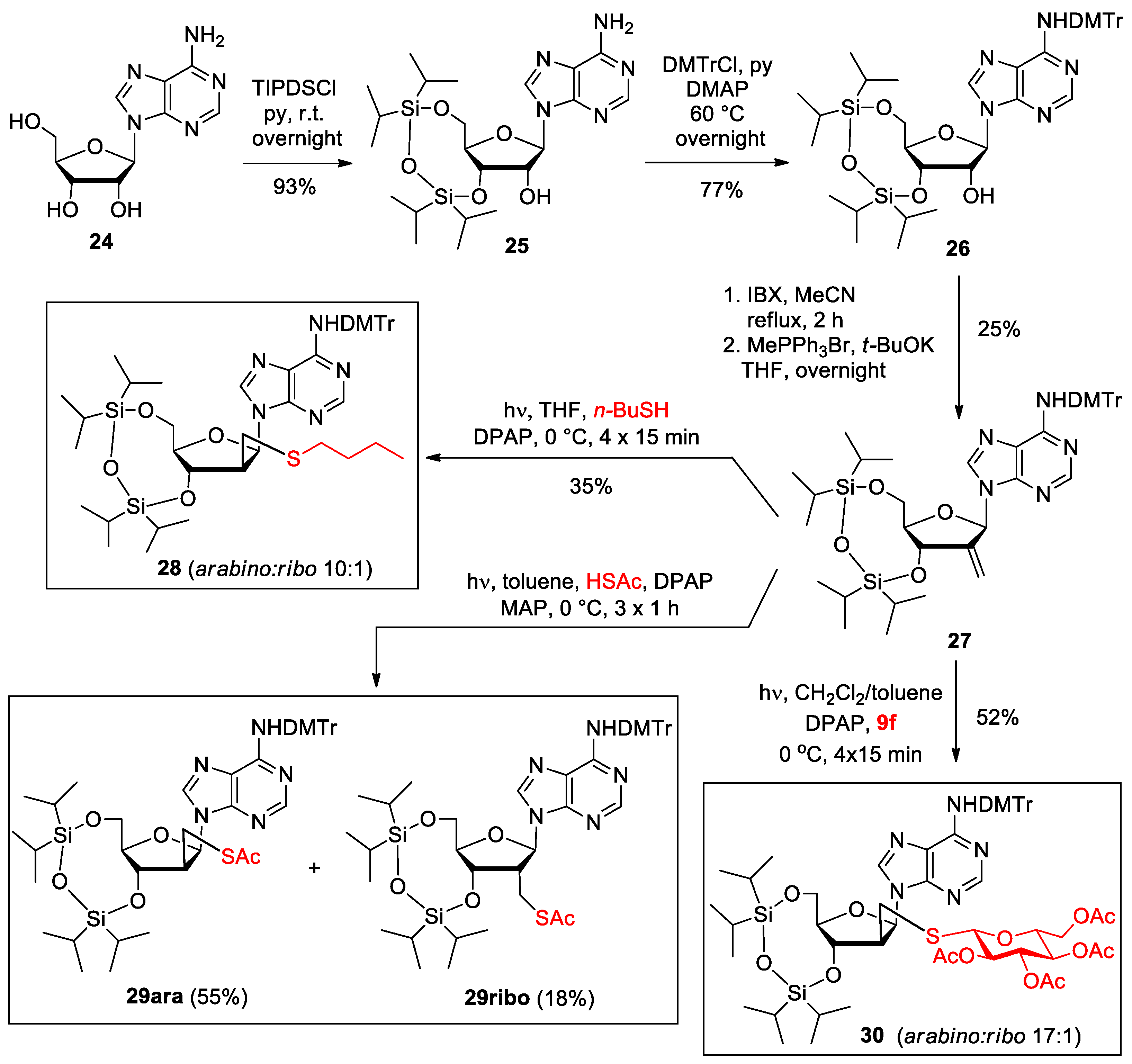
2.1. Synthesis
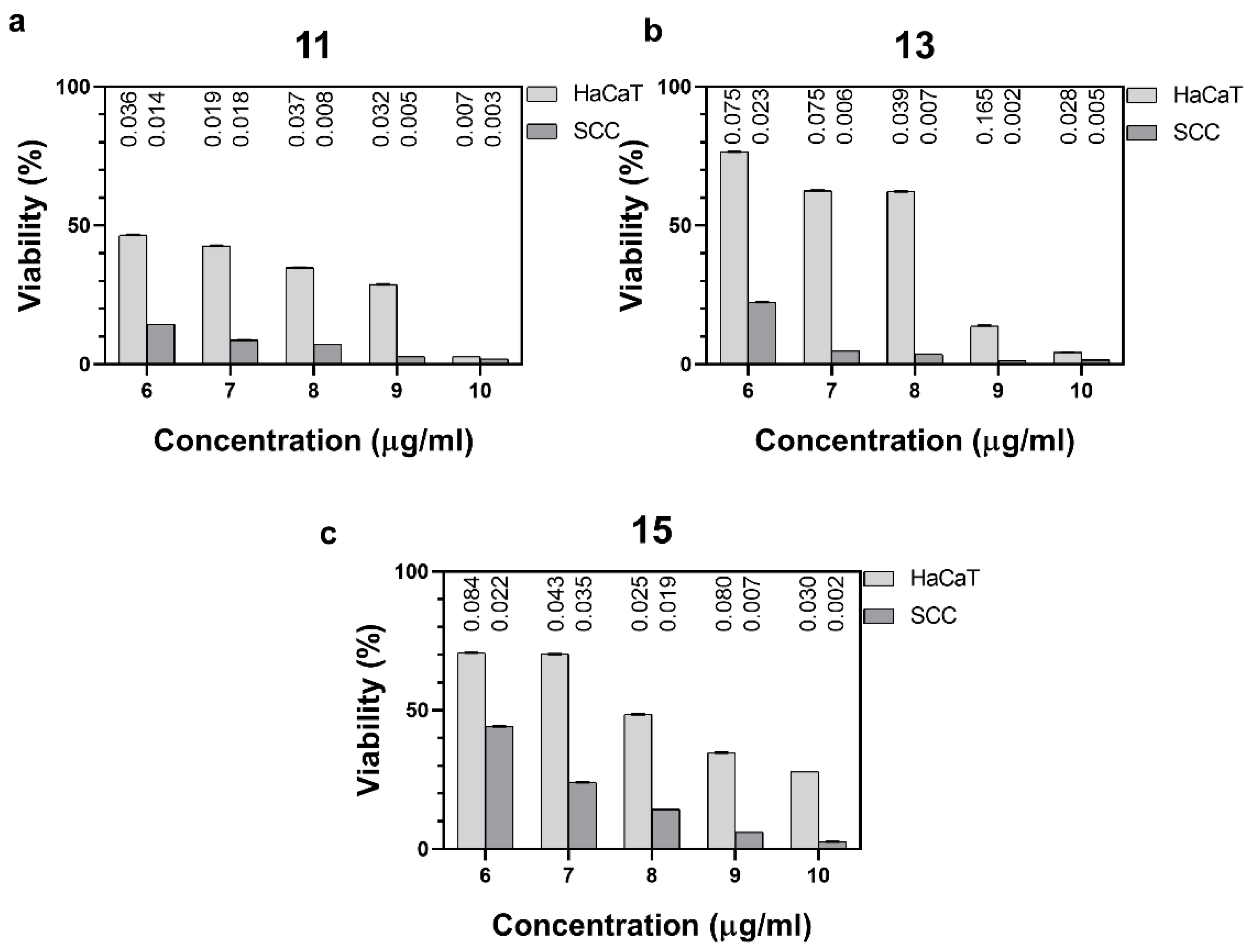
2.2. Cytotoxic Activity
2.3. Live-Cell Imaging via Time-Lapse Microscopy
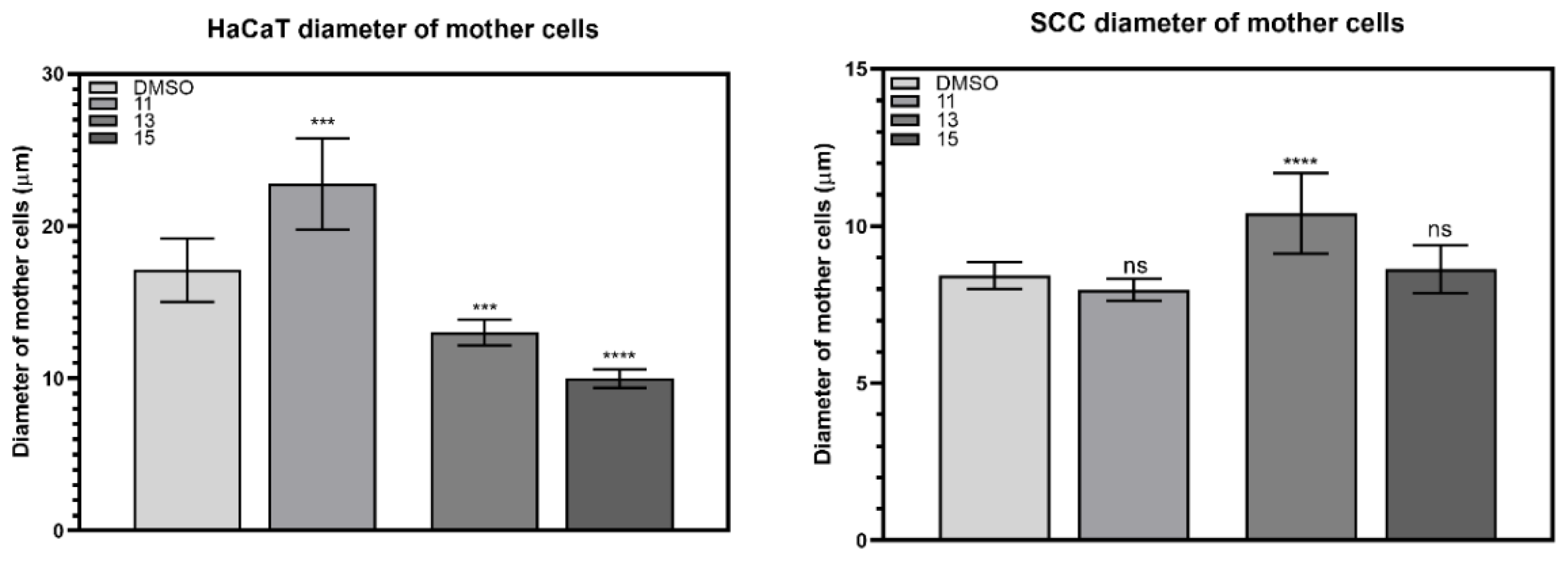
2.3.1. Changes in the Size of Mother Cells
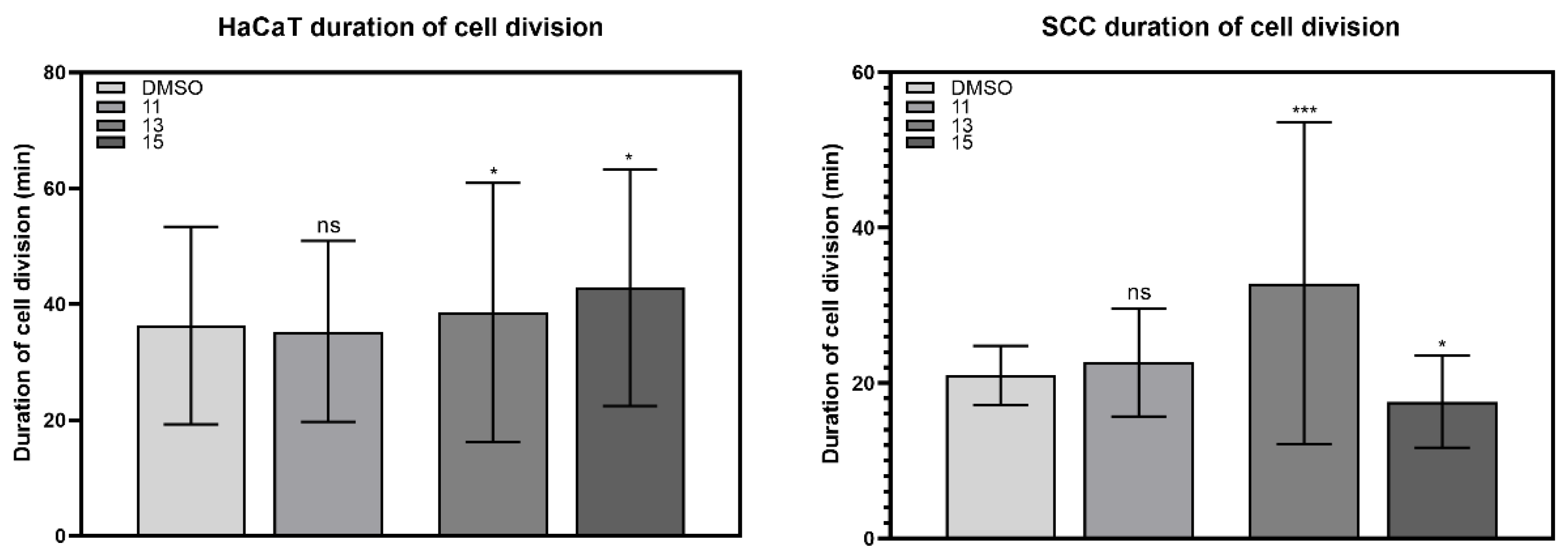
2.3.2. Duration of Cell Division
2.3.3. Generation Time
2.4. Antiviral Evaluation
3. Methods and Materials
3.1. Chemistry
3.1.1. 2′-Deoxy-2′-C-propylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-uracil (10)
3.1.2. 2′-Deoxy-2′-C-butylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-uracil (11)
3.1.3. 2′-Deoxy-2′-C-hexylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-uracil (12)
3.1.4. 2′-Deoxy-2′-C-acetylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-uracil (13)
3.1.5. 2′-Deoxy-2′-C-propylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-thymine (14)
3.1.6. 2′-Deoxy-2′-C-butylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-thymine (15)
3.1.7. 2′-Deoxy-2′-C-(5′-deoxyuridine-5′-yl)-sulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-thymine (16)
3.1.8. 2′-Deoxy-2′-C-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosylthio)methyl]-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-thymine (17ara) and 2′-deoxy-2′-C-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosylthio)methyl]-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl-thymine (17ribo)
3.1.9. 2′-Deoxy-2′-C-[(2-acetamido-3,4,6-tri-O-acetyl-2deoxy-β-D-glucopyranosylthio)methyl]-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl-thymine (18ara) and 2′-deoxy-2′-C-[(2-acetamido-3,4,6-tri-O-acetyl-2deoxy-β-D-glucopyranosylthio)methyl]-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-ribofuranosyl-thymine (18ribo)
3.1.10. 1-[2′-Deoxy-2′-C-(n-butylsulfanylmethyl)-β-D-arabinofuranosyl]-thymine (19)
3.1.11. Methyl 2-deoxy-2-C-methylene-3,5-O-(1,1,3,3-tetraisopropyl-l,3-disiloxanediyl)-β-D-erythro-pentofuranoside (22)
3.1.12. Methyl 2-C-(n-butylsulfanylmethyl)-2-deoxy-3,5-O-(1,1,3,3-tetraisopropyl-l,3-disiloxanediyl)-β-D-ribofuranoside (23)
3.1.13. 3′,5′-O-(1,1,3,3-Tetraisopropyldisiloxyl)-N-dimethoxytrityl-adenosine (26) [38]
3.1.14. 2′-Deoxy-N-dimethoxytrityl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxyl)--2′-C-methylene-adenosine (27)
3.1.15. [2′-Deoxy-2′-C-butylsulfanylmethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)]-D-arabinofuranosyl-N-dimethoxytrityl-adenine (28)
3.1.16. [2′-Deoxy-2′-C-acetylthiomethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)]-D-arabinofuranosyl-N-dimethoxytrityl-adenine (29ara) and [2′-deoxy-2′-C-acetylthiomethyl-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)]-adenosine (29ribo)
3.1.17. [2′-Deoxy-2′-C-[(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosylthio)methyl]-3′,5′-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)]-D-arabinofuranosyl-N-dimethoxytrityl-adenine (30)
3.2. Cell Viability Studies
MTT Assay
3.3. Time-Lapse Image Video-Microscopy and Image Analysis
3.3.1. Cell Size Measurements
3.3.2. Determination of the Time of Cell Division
3.3.3. Determination of Generation Time
3.4. Antiviral Assays
3.4.1. SARS-CoV-2 Infection, Cytopathic Effect (CPE) Detection, Compound Screening
3.4.2. Cytotoxicity in VERO E6 cells (XTT Assay CC50 Measurement)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogues. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Anglada, M.; Felipe, A.; Casado, F.J. Transport and mode of action of nucleoside derivatives used in chemical and antiviral therapies. Trends Pharmacol. Sci. 1998, 19, 424–430. [Google Scholar] [CrossRef]
- Thornton, P.J.; Kadri, H.; Miccoli, A.; Mehellou, Y. Nucleoside phosphate and phosphonate prodrug clinical candidates. J. Med. Chem. 2016, 59, 10400–10410. [Google Scholar] [CrossRef] [PubMed]
- Eyer, L.; Nencka, R.; de Clercq, E.; Seley-Radtke, K.L.; Růžek, D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir. Chem. Chemother. 2018, 26, 2040206618761299. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part I: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of aniviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef]
- Larson, R.A. Three New Drugs for Acute Lymphoblastic Leukemia: Nelarabine, Clofarabine, and Forodesine. Semin. Oncol. 2007, 34, S13–S20. [Google Scholar] [CrossRef]
- Jacobs, A.D. Gemcitabine-Based Therapy in Pancreas Cancer. Gemcitabine-Docetaxel and Other Novel Combinations. Cancer 2002, 95, 923–927. [Google Scholar] [CrossRef]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H.; Ding, X.; Svarovskaia, E.; Symonds, W.T.; Hindes, R.G.; Berrey, M.M. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. Recent applications of thiol–ene coupling as a click process for glycoconjugation. Chem. Soc. Rev. 2012, 41, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Lázár, L.; Csávás, M.; Herczeg, M.; Herczegh, P.; Borbás, A. Synthesis of S-Linked Glycoconjugates and S-Dissacharides by Thiol-Ene Coupling Reaction of Enoses. Org. Lett. 2012, 14, 4650–4653. [Google Scholar] [CrossRef] [PubMed]
- Lázár, L.; Csávás, M.; Tóth, M.; Somsák, L.; Borbás, A. Thio-click approach to the synthesis of stable glycomymetics. Chem. Pap. 2015, 69, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Bege, M.; Bereczki, I.; Herczeg, M.; Kicsák, M.; Eszenyi, D.; Herczegh, P.; Borbás, A. A low-temperature photoinduced thiol-ene click reaction: A method for the synthesis of sugar modified nucleosides. Org. Biomol. Chem. 2017, 15, 9226–9233. [Google Scholar] [CrossRef] [Green Version]
- Bege, M.; Kiss, A.; Kicsák, M.; Bereczki, I.; Baksa, V.; Király, G.; Szemán-Nagy, G.; Szigeti, M.Z.; Herczegh, P.; Borbás, A. Synthesis and cytostatic effect of 3’-deoxy-3’-C-sulfanylmethyl nucleoside derivatives with D-xylo configuration. Molecules 2019, 24, 2173. [Google Scholar] [CrossRef] [Green Version]
- Bege, M.; Bereczki, I.; Molnár, D.J.; Kicsák, M.; Pénzes-Daku, K.; Bereczky, Z.; Ferenc, G.Y.; Kovács, L.; Herczegh, P.; Borbás, A. Synthesis and oligomerization of cysteinyl nucleosides. Org. Biomol. Chem. 2020, 18, 8161–8178. [Google Scholar] [CrossRef]
- Borbás, A. Photoinitiated Thiol-ene Reactions of Enoses: A Powerful Tool for Stereoselective Synthesis of Glycomimetics with Challenging Glycosidic Linkages. Chem. Eur. J. 2020, 26, 6090–6101. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.; Baksa, V.; Bege, M.; Tálas, L.; Borbás, A.; Bereczki, I.; Bánfalvi, G.; Szemán-Nagy, G. MTT test and time-lapse microscopy to evaluate the antitumor potential of nucleoside analogues. Anticancer Res. 2021, 41, 137–149. [Google Scholar] [CrossRef]
- Hodek, J.; Veselovská, L.; Sýkorová, V.; Cízék, K.; Pohl, R.; Eyer, L.; Svoboda, P.; Ruzek, D.; Weber, J.; Nencka, R.; et al. Antiviral Activity of 7-Substituted 7-Deazapurine Ribonucleosides, Monophosphate Prodrugs, and Triphoshates against Emerging RNA Viruses. ACS Infect. Dis. 2021, 7, 471–478. [Google Scholar]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; André, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antivir. Res. 2022, 198, 105252. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, F.; Malher, A.; Vasseur, J.-J.; Dupouy, C.; Debart, F. Conjugation of Small Molecules to RNA Using a Reducible Disulfide Linker Attached at the 2′-OH Position through a Carbamate Function. Eur. J. Org. Chem. 2019, 2019, 5636–5645. [Google Scholar] [CrossRef]
- Masaki, Y.; Yamamoto, K.; Inde, T.; Yoshida, K.; Maruyama, A.; Nagata, T.; Tanihata, J.; Takeda, S.; Sekine, M.; Seioa, K. Synthesis of 2′-O-(N-methylcarbamoylethyl) 5-methyl-2-thiouridine and its application to splice-switching oligonucleotides. Bioorg. Med. Chem. Lett. 2019, 29, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-S.; Luo, M.-Z.; Liu, M.-C.; Clarke-Katzenburg, R.H.; Cheng, Y.-C.; Prusoff, W.H.; Mancini, W.R.; Birnbaum, G.I.; Gabe, E.J.; Giziewicz, J. Synthesis and Anticancer and Antiviral Activities of Various 2′- and 3′-Methylidene-Substituted Nucleoside Analogues and Crystal Structure of 2′-Deoxy-2′-methylidenecytidine Hydrochloride. J. Med. Chem. 1991, 34, 2607–2615. [Google Scholar] [CrossRef]
- Reist, E.J.; Benitez, A.; Goodman, L. The Synthesis of Some S’-Thiopentofuranosylpyrimidines. J. Chem. Soc. 1964, 29, 554–558. [Google Scholar] [CrossRef]
- Dénès, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl radicals in organic synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef]
- Buchini, S.; Leumann, C.J. New Nucleoside Analogues for the Recognition of Pyrimidine-Purine Inversion Sites. Nucl. Nucl. Nucleic Acids 2003, 22, 1199–1201. [Google Scholar] [CrossRef]
- Bockman, M.R.; Kalinda, A.S.; Petrelli, R.; De la Mora-Rey, T.; Tiwari, D.; Liu, F.; Dawadi, S.; Nandakumar, M.; Rhee, K.Y.; Schnappinger, D.; et al. Targeting Mycobacterium tuberculosis Biotin Protein Ligase (MtBPL) with Nucleoside-Based Bisubstrate Adenylation Inhibitors. J. Med. Chem. 2015, 58, 7349–7369. [Google Scholar] [CrossRef] [Green Version]
- Kelemen, V.; Bege, M.; Eszenyi, D.; Debreczeni, N.; Bényei, A.; Stürzer, T.; Herczegh, P.; Borbás, A. Stereoselective Thioconjugation by Photoinduced Thiol-ene Coupling Reactions of Hexo-and Pentopyranosyl D-and L-Glycals at Low-Temperature—Reactivity and Stereoselectivity Study. Chem. Eur. J. 2019, 25, 14477. [Google Scholar] [CrossRef]
- Le, S.T.; Páll, D.; Rőth, E.; Tran, T.; Debreczeni, N.; Bege, M.; Bereczki, I.; Ostorházi, E.; Milánkovits, M.; Herczegh, P.; et al. The very first modification of pleuromutilin and lefamulin by photoinitiated radical addition reactions—Synthesis and antibacterial studies. Pharmaceutics 2021, 13, 2028. [Google Scholar] [CrossRef]
- McCourt, R.O.; Scanlan, E.M. A sequential acyl thiol–ene and thiolactonization approach for the synthesis of δ-thiolactones. Org. Lett. 2019, 21, 3460–3464. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, Á.; Fenyvesi, F.; Majercsik, O.; Pelyvás, I.F.; Bácskay, I.; Fehér, P.; Váradi, J.; Vecsernyés, M.; Herczegh, P. Synthesis and Cytotoxicity of Leinamycin Antibiotic Analogues. J. Med. Chem. 2006, 49, 5626–5630. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.A.; Oliveira, M.; Christiansen, M.A.; Cutler, C.E. Preliminary SAR analysis of novel antiproliferative N6,5’-bis-ureidoadenosine derivatives. Bioorg. Med. Chem. Lett. 2009, 19, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.R.; Cutler, C.E.; Browning, M.S.; Balzarini, J.; Peterson, M.A. Synthesis and SAR of 2’,3’-bis-O-substituted N6,5’-bis-ureidoadenosine derivatives: Implications for prodrug delivery and mechanism of action. Bioorg. Med. Chem. Lett. 2012, 22, 6067–6071. [Google Scholar] [CrossRef]
- Chamorro, C.; Pérez-Pérez, M.J.; Rodríguez-Barrios, F.; Gago, F.; De Clercq, E.; Balzarini, J.; San-Félix, A.; Camarasa, M.J. Exploring the role of the 5’-position of TSAO-T. Synthesis and anti-HIV evaluation of novel TSAO-T derivatives. Antivir. Res. 2001, 50, 207–222. [Google Scholar] [CrossRef]
- Harmse, L.; Dahan-Farkas, N.; Panayides, J.L.; van Otterlo, W.; Penny, C. Aberrant Apoptotic Response of Colorectal Cancer Cells to Novel Nucleoside Analogues. PLoS ONE 2015, 10, e0138607. [Google Scholar] [CrossRef] [Green Version]
- Panayides, J.L.; Mathieu, V.; Banuls, L.M.Y.; Apostolellis, H.; Dahan-Farkas, N.; Davids de Leonie Harmse, H.; Rey, M.E.C.; Green, I.R.; Pelly, S.C.; Kiss, R.; et al. Synthesis and in vitro growth inhibitory activity of novel silyl- and trityl-modified nucleosides. Bioorg. Med. Chem. 2016, 24, 2716–2724. [Google Scholar] [CrossRef]
- Zhong, M.; Strobel, S.A.A. Synthesis of isotopically labeled P-site substrates for the ribosomal peptidyl transferase reaction. J. Org. Chem. 2008, 73, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Boukamp, P.; Popp, S.; Altmeyer, S.; Hülsen, A.; Fasching, C.; Cremer, T.; Fusenig, N.E. Sustained nontumorigenic phenotype correlates with a largely stable chromosome content during long-term culture of the human keratinocyte line HaCaT. Genes Chromosom. Cancer 1997, 19, 201–214. [Google Scholar] [CrossRef]
- Sominski, D.D.; Rafferty, P.; Brosnan, K.; Volk, A.; Walker, M.; Capaldi, D.; Emmell, E.; Johnson, K.; Weinstock, D. Development of a squamous cell carcinoma mouse model for immunotoxicity testing. J. Immunotoxicol. 2016, 13, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Király, G.; Veres, P.; Lázár, I.; Fábián, I.; Bánfalvi, G.; Juhász, I.; Kalmár, J. Controlled release of methotrexate from functionalized silica-gelatin aerogel microparticles applied against tumour cell growth. Int. J. Pharm. 2019, 558, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, J.; Vesely, P.; Netíková, I.; Matousková, E.; Petruzelka, L. The protective effect of pyrimidine nucleosides on human HaCaT keratinocytes treated with 5-FU. Anticancer Res. 2015, 35, 1303–1310. [Google Scholar]
- Nagy, Z.; Nagy, M.; Kiss, A.; Rácz, D.; Barna, B.; Könczöl, P.; Bankó, C.; Bacsó, Z.; Kéki, S.; Bánfalvi, G.; et al. MICAN, a new fluorophore for vital and non-vital staining of human cells. Toxicol. In Vitro 2018, 48, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Hennig, G.W.; Petrenyi, K.; Kovacs, L.; Pocsi, I.; Dombradi, V.; Bánfalvi, G. Time-lapse video microscopy and image analysis of adherence and growth patterns of Candida albicans strains. Appl. Microbiol. Biotechnol. 2014, 98, 5185–5194. [Google Scholar] [CrossRef]
- Farkas, E.; Ujvarosi, K.; Nagy, G.; Posta, J.; Bánfalvi, G. Apoptogenic and necrogenic effects of mercuric acetate on the chromatin structure of K562 human erythroleukemia cells. Toxicol. In Vitro 2010, 24, 267–275. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkene | R’ | Temperature | Product | arabino:ribob | Yield |
|---|---|---|---|---|---|
| 7 | n-propyl | 0 °C | 10 | 14:1 | 65% |
| 7 | n-butyl | 0 °C | 11 | 20:1 | 69% |
| 7 | n-hexyl | 0 °C | 12 | 9:1 | 58% |
| 7 | acetyl | 0 °C | 13 | 8:1 | 57% |
| 8 | n-propyl | 0 °C | 14 | 20:1 | 74% |
| 8 | n-butyl | 0 °C | 15 | 13:1 | 67% |
| 8 | uridine-5′-yl c | 0 °C | 16 | 7:1 | 42% c |
| 8 | Glc (OAc) 4 | 0 °C | 17 | 1.1:1 | 67% |
| 8 | Glc (OAc) 4 | −80 °C | 17 | 1.4:1 | 69% |
| 8 | GlcNAc(OAc) 3 | 0 °C | 18 | 2.3:1 | 60% |
| Compound | HaCaT IC50 (µM) | SCC-VII IC50 (µM) | SI a |
|---|---|---|---|
| 2a b | 17.5 | 9.8 | 1.8 |
| 2b b | n.a. | n.a. | |
| 6 | 20.3 ± 0.94 | 15.4 ± 0.53 | 1.3 |
| 8 | 18.8 ± 0.68 | 13.3 ± 0.20 | 1.4 |
| 10 | 8.0 ± 0.25 | 10.9 ± 0.03 | 0.7 |
| 11 | 12.1 ± 0.19 | 3.7 ± 0.39 | 3.3 |
| 12 | 15.5 ± 0.25 | 12.4 ± 0.21 | 1.3 |
| 13 | 13.5 ± 0.11 | 4.7 ± 0.24 | 2.9 |
| 14 | 18.8 ± 0.09 | 16.0 ± 0.37 | 1.2 |
| 15 | 13.7 ± 0.16 | 7.5 ± 0.12 | 1.8 |
| 16 | 17.7 ± 2.04 | 45.7 ± 0.54 | 0.4 |
| 19 | n.a.c | n.a. | |
| 23 | n.a. | n.a. | |
| 27 | n.a. | n.a. | |
| 28 | 29.4 ± 5.91 | 45.3 ± 6.21 | 0.6 |
| 29ara | n.a. | 28.2 ± 8.40 | |
| 29ribo | n.a. | n.a. | |
| 30 | n.a. | n.a. | |
| 5-FUd | - | 60.7 ± 8.45 |
| Change in Cell Sizes before Division (%) b | |||
|---|---|---|---|
| 11 | 13 | 15 | |
| HaCaT n = 30 | 29.23 ± 0.60 | −23.90 ± 0.83 | −41.57 ± 0.61 |
| SCC VII n = 30 | −5.34 ± 0.35 | 23.36 ± 1.27 | 2.37 ± 0.76 |
| Difference in the Duration of Cell Division (min) Relative to the Control in (%) b | |||
|---|---|---|---|
| 11 | 13 | 15 | |
| HaCaT n = 30 | −2.75 ± 15.62 | 6.25 ± 22.36 | 18.04 ± 20.45 |
| SCC VII n = 30 | 8.00 ± 3.82 | 56.55 ± 20.70 | −16.06 ± 5.92 |
| Difference in the Duration of Cell Cycle (h) Change in % b | |||
|---|---|---|---|
| 11 | 13 | 15 | |
| HaCaT n = 30 | 29.10 ± 2.57 | 32.96 ± 3.00 | 27.41 ± 2.83 |
| SCC VII n = 30 | 20.40 ± 2.97 | 12.86 ± 3.72 | 30.00 ± 3.06 |
| Compound | SARS-CoV-2 in VERO E6 Cells | HCoV-229E in HEL Cells | |||
|---|---|---|---|---|---|
| EC50 CPE (µM) | 95% CI of EC50 CPE | CC50 (µM) | EC50 MTS (µM) | CC50 (µM) | |
| 2a | >35 | n.a. | ~35 | n.d. | n.d. |
| 2b | n.d. | n.d. | n.d. | 7.4 | 14 |
| 8 | n.d. | n.d. | n.d. | 4.0 | 10 |
| 10 | >16 | n.a. | 15 | n.d. | n.d. |
| 11 | >6.4 | n.a. | ~6.4–16 | 4.1 | 10 |
| 12 | ~13 | very wide | ~18 | n.d. | n.d. |
| 13 | >100 | n.a. | >100 | 5.7 | 10 |
| 15 | 17 | 9.7–29 | 16.6 | n.d. | n.d. |
| 16 | >100 | n.a. | >100 | >100 | 11 |
| 17 | >100 | n.a. | >100 | 4.9 | 9.2 |
| 18 | n.d. | n.d. | n.d. | >100 | 10 |
| 19 | >100 | n.a. | >100 | >100 | >100 |
| 28 | >100 | n.a. | >100 | >100 | >100 |
| 29ara | n.d. | n.d. | n.d. | >100 | >100 |
| 29ribo | n.d. | n.d. | n.d. | >100 | >100 |
| 30 | >100 | n.a. | >100 | >100 | >100 |
| Remdesivir | 2.2 | 1.7 to 2.8 | >100 | n.d. | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bege, M.; Kiss, A.; Bereczki, I.; Hodek, J.; Polyák, L.; Szemán-Nagy, G.; Naesens, L.; Weber, J.; Borbás, A. Synthesis and Anticancer and Antiviral Activities of C-2′-Branched Arabinonucleosides. Int. J. Mol. Sci. 2022, 23, 12566. https://doi.org/10.3390/ijms232012566
Bege M, Kiss A, Bereczki I, Hodek J, Polyák L, Szemán-Nagy G, Naesens L, Weber J, Borbás A. Synthesis and Anticancer and Antiviral Activities of C-2′-Branched Arabinonucleosides. International Journal of Molecular Sciences. 2022; 23(20):12566. https://doi.org/10.3390/ijms232012566
Chicago/Turabian StyleBege, Miklós, Alexandra Kiss, Ilona Bereczki, Jan Hodek, Lenke Polyák, Gábor Szemán-Nagy, Lieve Naesens, Jan Weber, and Anikó Borbás. 2022. "Synthesis and Anticancer and Antiviral Activities of C-2′-Branched Arabinonucleosides" International Journal of Molecular Sciences 23, no. 20: 12566. https://doi.org/10.3390/ijms232012566