Study of Biomolecular Interactions of Mitochondrial Proteins Related to Alzheimer’s Disease: Toward Multi-Interaction Biomolecular Processes


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Surface Plasmon Resonance (SPR) Biosensor
2.3. Preparation of Aβ
2.4. Characterization of Oligomerization State of Aβ
2.5. Characterization of cypD and 17β-HSD10
2.6. Determination of Kinetic Parameters
3. Results and Discussion
3.1. Characterization of Oligomerization State of Aβ
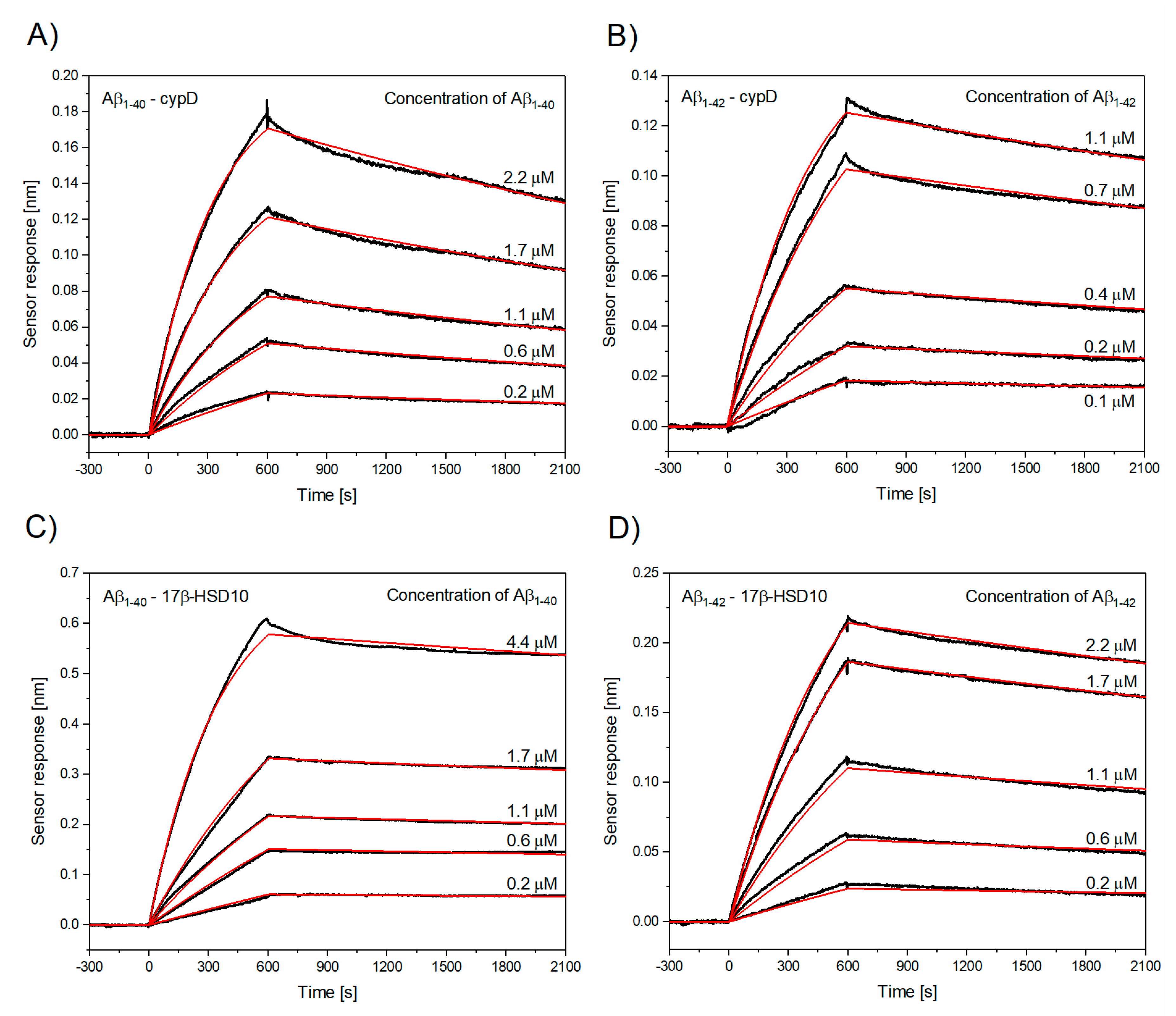
3.2. Determination of Kinetic Parameters of the Interactions between Aβ and cypD or 17β-HSD10
3.3. Modeling the Complex Biomolecular Interaction Interplay in Mitochondria
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fa, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-beta and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. JAD 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Yan, S.S. Synaptic Mitochondrial Pathology in Alzheimer’s Disease. Antioxid. Redox Signal. 2012, 16, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Suzuki, N.; Cheung, T.; Cai, X.; Odaka, A.; Otvos, L.; Eckman, C.; Golde, T.; Younkin, S. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid β peptides. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. JAD 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Zhou, H.-D.; Zhou, X.-F. Clearance of amyloid-beta in Alzheimer’s disease: Progress, problems and perspectives. Drug Discov. Today 2006, 11, 931–938. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Crouch, P.J.; Harding, S.-M.E.; White, A.R.; Camakaris, J.; Bush, A.I.; Masters, C.L. Mechanisms of Aβ mediated neurodegeneration in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2008, 40, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garai, K.; Frieden, C. Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ. Proc. Natl. Acad. Sci. USA 2013, 110, 3321–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Wang, C. Aβ42 is More Rigid than Aβ40 at the C Terminus: Implications for Aβ Aggregation and Toxicity. J. Mol. Biol. 2006, 364, 853–862. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Benek, O.; Aitken, L.; Hroch, L.; Kuca, K.; Gunn-Moore, F.; Musilek, K. A Direct Interaction between Mitochondrial Proteins and Amyloid-beta Peptide and its Significance for the Progression and Treatment of Alzheimer’s Disease. Curr. Med. Chem. 2015, 22, 1056–1085. [Google Scholar] [CrossRef] [Green Version]
- Muirhead, K.E.; Borger, E.; Aitken, L.; Conway, S.J.; Gunn-Moore, F.J. The consequences of mitochondrial amyloid beta-peptide in Alzheimer’s disease. Biochem. J. 2010, 426, 255–270. [Google Scholar] [CrossRef]
- Pagani, L.; Eckert, A. Amyloid-Beta interaction with mitochondria. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Readnower, R.D.; Sauerbeck, A.D.; Sullivan, P.G. Mitochondria, Amyloid β, and Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.D.; Stern, D.M. Mitochondrial dysfunction and Alzheimer’s disease: Role of amyloid-β peptide alcohol dehydrogenase (ABAD). Int. J. Exp. Pathol. 2005, 86, 161–171. [Google Scholar] [CrossRef]
- Singh, P.; Suman, S.; Chandna, S.; Das, T.K. Possible role of amyloid-beta, adenine nucleotide translocase and cyclophilin-D interaction in mitochondrial dysfunction of Alzheimer’s disease. Bioinformation 2009, 3, 440–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflügers Arch. Eur. J. Physiol. 2010, 459, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Yan, S.; Fu, J.; Soto, C.; Chen, X.; Zhu, H.; Al-Mohanna, F.; Collison, K.; Zhu, A.; Stern, E.; Saido, T.; et al. An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer’s disease. Nature 1997, 389, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Du Yan, S.; Shi, Y.; Zhu, A.; Fu, J.; Zhu, H.; Zhu, Y.; Gibson, L.; Stern, E.; Collison, K.; Al-Mohanna, F.; et al. Role of ERAB/l-3-Hydroxyacyl-coenzyme A Dehydrogenase Type II Activity in Aβ-induced Cytotoxicity. J. Biol. Chem. 1999, 274, 2145–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aß to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Hemmerová, E.; Špringer, T.; Krištofiková, Z.; Homola, J. In vitro study of interaction of 17β-hydroxysteroid dehydrogenase type 10 and cyclophilin D and its potential implications for Alzheimer’s disease. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Liu, Y.; Sorci, M.; Belfort, G.; Lustbader, J.W.; Yan, S.S.; Wang, C. Surface Plasmon Resonance and Nuclear Magnetic Resonance Studies of ABAD−Aβ Interaction. Biochemistry 2007, 46, 1724–1731. [Google Scholar] [CrossRef]
- Aitken, L.; Quinn, S.D.; Perez-Gonzalez, D.C.; Samuel, I.D.W.; Penedo-Esteiro, J.C.; Gunn-Moore, F.J. Morphology-specific inhibition of β-amyloid aggregates by 17β-hydroxysteroid dehydrogenase type 10. ChemBioChem 2016, 17, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Špringer, T.; Piliarik, M.; Homola, J. Surface plasmon resonance sensor with dispersionless microfluidics for direct detection of nucleic acids at the low femtomole level. Sens. Actuators B Chem. 2010, 145, 588–591. [Google Scholar] [CrossRef]
- Špringer, T.; Chadtová Song, X.; Ermini, M.L.; Lamačová, J.; Homola, J. Functional gold nanoparticles for optical affinity biosensing. Anal. Bioanal. Chem. 2017, 409, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Shao, H.; Zhang, Y.; Li, H.; Menon, N.K.; Neuhaus, E.B.; Brewer, J.M.; Byeon, I.-J.L.; Ray, D.G.; Vitek, M.P.; et al. Solution NMR Studies of the Aβ(1−40) and Aβ(1−42) Peptides Establish that the Met35 Oxidation State Affects the Mechanism of Amyloid Formation. J. Am. Chem. Soc. 2004, 126, 1992–2005. [Google Scholar] [CrossRef] [PubMed]
- Broersen, K.; Jonckheere, W.; Rozenski, J.; Vandersteen, A.; Pauwels, K.; Pastore, A.; Rousseau, F.; Schymkowitz, J. A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng. Des. Sel. 2011, 24, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, M.; Naldi, M.; Fiori, J.; Valle, F.; Biscarini, F.; Nicolau, D.V.; Andrisano, V. Kinetic characterization of amyloid-beta 1–42 aggregation with a multimethodological approach. Anal. Biochem. 2011, 414, 215–225. [Google Scholar] [CrossRef]
- Bruggink, K.A.; Muller, M.; Kuiperij, H.B.; Verbeek, M.M. Methods for analysis of amyloid-beta aggregates. J. Alzheimer’s Dis. JAD 2012, 28, 735–758. [Google Scholar] [CrossRef]
- Aguilar, M.-I.; Small, D.H. Surface plasmon resonance for the analysis of β-amyloid interactions and fibril formation in alzheimer’s disease research. Neurotox. Res. 2005, 7, 17–27. [Google Scholar] [CrossRef]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar] [CrossRef]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 1247–1257. [Google Scholar] [CrossRef]
- Bradshaw, P.C.; Pfeiffer, D.R. Release of Ca2+ and Mg2+ from yeast mitochondria is stimulated by increased ionic strength. BMC Biochem. 2006, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gout, E.; Rebeille, F.; Douce, R.; Bligny, R. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: Unravelling the role of Mg2+ in cell respiration. Proc. Natl. Acad. Sci. USA 2014, 111, E4560–E4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Anton Merwe, P.; Neil Barclay, A. Transient intercellular adhesion: The importance of weak protein-protein interactions. Trends Biochem. Sci. 1994, 19, 354–358. [Google Scholar] [CrossRef]
- Krištofiková, Z.; Špringer, T.; Gedeonová, E.; Hofmannová, A.; Říčný, J.; Hromadková, L.; Vyhnálek, M.; Laczo, J.; Nikolai, T.; Hort, J.; et al. Interactions of 17β-Hydroxysteroid Dehydrogenase Type 10 and Cyclophilin D in Alzheimer’s Disease. Neurochem. Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Interaction | ka [M−1s−1] | kd [s−1] | KD [nM] |
|---|---|---|---|
| Aβ1–40–cypD | (1.17 ± 0.15) × 103 | (1.74 ± 0.57) × 10−4 | 160.2 ± 57.8 |
| Aβ1–42–cypD | (2.69 ± 1.22) × 103 | (1.39 ± 0.41) × 10−4 | 56.5 ± 5.4 |
| Aβ1–40–17β-HSD10 | (0.63 ± 0.05) × 103 | (0.47 ± 0.04) × 10−4 | 74.5 ± 1.8 |
| Aβ1–42–17β-HSD10 | (0.65 ± 0.25) × 103 | (1.12 ± 0.52) × 10−4 | 181.4 ± 16.0 |
| The Interaction | ka [M−1s−1] | kd [s−1] | KD [nM] |
|---|---|---|---|
| Oligomeric Aβ1–42–cypD | (11.12 ± 1.09) × 103 | (0.61 ± 0.12) × 10−4 | 5.3 ± 1.2 |
| Oligomeric Aβ1–42–17β-HSD10 | (4.04 ± 0.62) × 103 | (0.31 ± 0.09) × 10−4 | 8.0 ± 3.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmerová, E.; Špringer, T.; Krištofiková, Z.; Homola, J. Study of Biomolecular Interactions of Mitochondrial Proteins Related to Alzheimer’s Disease: Toward Multi-Interaction Biomolecular Processes. Biomolecules 2020, 10, 1214. https://doi.org/10.3390/biom10091214
Hemmerová E, Špringer T, Krištofiková Z, Homola J. Study of Biomolecular Interactions of Mitochondrial Proteins Related to Alzheimer’s Disease: Toward Multi-Interaction Biomolecular Processes. Biomolecules. 2020; 10(9):1214. https://doi.org/10.3390/biom10091214
Chicago/Turabian StyleHemmerová, Erika, Tomáš Špringer, Zdeňka Krištofiková, and Jiří Homola. 2020. "Study of Biomolecular Interactions of Mitochondrial Proteins Related to Alzheimer’s Disease: Toward Multi-Interaction Biomolecular Processes" Biomolecules 10, no. 9: 1214. https://doi.org/10.3390/biom10091214