Corrigendum: Cysteine residues in signal transduction and its relevance in pancreatic beta cells
 Blanka Holendova
Blanka Holendova Lydie Plecita-Hlavata
Lydie Plecita-Hlavata- Laboratory of Pancreatic Islet Research, Institute of Physiology, Czech Academy of Sciences, Prague, Czechia
Cysteine is one of the least abundant but most conserved amino acid residues in proteins, playing a role in their structure, metal binding, catalysis, and redox chemistry. Thiols present in cysteines can be modified by post-translational modifications like sulfenylation, acylation, or glutathionylation, regulating protein activity and function and serving as signals. Their modification depends on their position in the structure, surrounding amino acids, solvent accessibility, pH, etc. The most studied modifications are the redox modifications by reactive oxygen, nitrogen, and sulfur species, leading to reversible changes that serve as cell signals or irreversible changes indicating oxidative stress and cell damage. Selected antioxidants undergoing reversible oxidative modifications like peroxiredoxin-thioredoxin system are involved in a redox-relay signaling that can propagate to target proteins. Cysteine thiols can also be modified by acyl moieties’ addition (derived from lipid metabolism), resulting in protein functional modification or changes in protein anchoring in the membrane. In this review, we update the current knowledge on cysteine modifications and their consequences in pancreatic β-cells. Because β-cells exhibit well-balanced redox homeostasis, the redox modifications of cysteines here serve primarily for signaling purposes. Similarly, lipid metabolism provides regulatory intermediates that have been shown to be necessary in addition to redox modifications for proper β-cell function and, in particular, for efficient insulin secretion. On the contrary, the excess of reactive oxygen, nitrogen, and sulfur species and the imbalance of lipids under pathological conditions cause irreversible changes and contribute to oxidative stress leading to cell failure and the development of type 2 diabetes.
1 Introduction
Cysteine (Cys) exhibits unique character. It contains sulfur in the form of sulfhydryl thiol form which is ionizable, forming a negatively charged thiolate group after deprotonation that further increases its reactivity. This thiol/thiolate group is subject to alkylation by electrophiles and oxidation by reactive oxygen, nitrogen, and sulfur species, resulting in posttranslationally modified forms that may exhibit significantly altered functions (1).
Cys is considered one of the most conserved amino acids in proteins across all species. Strong selection pressure retained Cys residues at functionally important sites and removed them from others (2). The physical and chemical properties of Cys predestine it to be a polar residue (like serine, where the sulfur atom is replaced by oxygen), however, it is considered hydrophobic because it is buried to an increased extent (protection from the solvent), and the number of unpaired Cys residues on the protein surface is minimal (2). Another unique property is its tendency to form clusters with other Cys, as in metal-binding or redox-sensitive proteins, leading to the formation of disulfide bonds. Exposed and isolated Cys are less conserved (1).
2 Chemical properties of cysteine
The chemical properties of Cys allow it to be both redox active and strongly nucleophilic due to the large atomic radius of the sulfur atom, the presence of lone pairs of electrons, and the low dissociation energy of the thiol S-H bond.
The thiol groups (R- SH) undergo deprotonation (loss of H+), giving the thiolate form R-S-. The readiness to provide the proton is given by the pKa, the local pH and electrostatic environment. In proteins, the specific hydrogen bond donors and an electropositive local environment cause a decrease in the pKa value due to the stabilization of the negative thiolate anion, while a hydrophobic environment or an electronegative local environment has the opposite effect (1).
Another important feature is the reductive potential of the thiol group, i.e., the ability to accept or donate electrons. This ability allows the formation of so-called redox pairs, and their interconversion is defined as a reductive or oxidative half-reaction. In biological systems, electron transfer is very dynamic and involves many players. Therefore, many reactions occur even under thermodynamically unfavorable conditions and would never occur in isolation, favoring the kinetic pathways and rates associated with certain reactions (3, 4).
Thiols can produce disulfides in one or more thiol-disulfide exchange processes in intricate biological systems (1). Long assumed to merely serve to stabilize proteins structurally, it is now known that these processes also give rise to many enzymes’ diverse and dynamic functional characteristics (5). The rate-determining stage in the folding process of proteins creating structural disulfide linkages is direct thiol-disulfide exchange. Although in vivo enzyme catalysis speeds up the events, spontaneous thiol-disulfide exchange is slow (kinetically inadequate on the folding timescale). Through a sequence of intra- and intermolecular thiol-disulfide exchange processes carried out by oxidoreductases, the folding polypeptide gains a new disulfide bond. Molecular oxygen serves as the oxidizing equivalent in these reactions. A different mechanism for thiol-disulfide exchange involves the oxidative conversion of protein thiols to disulfides and their subsequent reduction. Protein thiols are also significant targets of reactive oxygen species in vivo. These processes are crucial for both antioxidant defense and redox regulation of cell signaling, and it is believed that the thiol-disulfide pool is principally responsible for maintaining intracellular redox equilibrium (6). It is now widely acknowledged that thiol-disulfide exchange and thiol oxidation/reduction reactions are dynamic, non-equilibrium processes that are kinetically rather than thermodynamically controlled in cellular systems (7–9). In other words, the partitioning of particular routes depends on relative rates, whereas redox potentials and equilibrium constants just tell whether a reaction is favorable. By fine-tuning the activation energies of reactions that control the outcome of oxidative stimuli or the location of structural disulfides in native proteins, enzymes play a crucial role in these processes.
3 Oxidative posttranslational modifications of cysteines
The availability of different oxidation states of thiol-sulfur allows the formation of a variety of oxidative posttranslational modifications (PTMs) on cysteines, including S-nitrosylation (or S-nitrosation, SNO), sulfhydration (SSH), disulfide bond formation (RS-SR), sulfenylation (SOH), sulfinic acid (SO2H), and sulfonic acid (SO3H) (10) (Figure 1A). Most Cys oxidative PTMs are initiated by reactive oxygen or nitrogen species (ROS/RNS) or sulfane (H2S) reacting with the free thiol on a Cys side chain (Figure 1A). Two-electron oxidation of thiol(ate) groups, e.g., by H2O2, peroxynitrite, and other hydroperoxides, produces the simplest oxyacid of sulfur, sulfenic acid. The rate of this reaction can vary from negligibly slow to 108 M-1 s-1 at active sites of peroxidase (4). Sulfenic acid readily reacts with proximal thiol groups to form disulfides or is further oxidized to irreversible sulfinic or sulfonic acids in the absence of such groups (11). Sulfonic acids can also react with each other to form thiosulfinates or with amine or amide groups to form sulfenylamides (12) (Figure 1A). S-nitrosothiols and persulfides are additional oxidative reaction byproducts that are unquestionably significant as signaling intermediates (13, 14). The thiyl radical, another reactive species that can produce a variety of products once generated, can be produced by the one-electron oxidation of thiol groups in the presence of radicals (15).
Figure 1
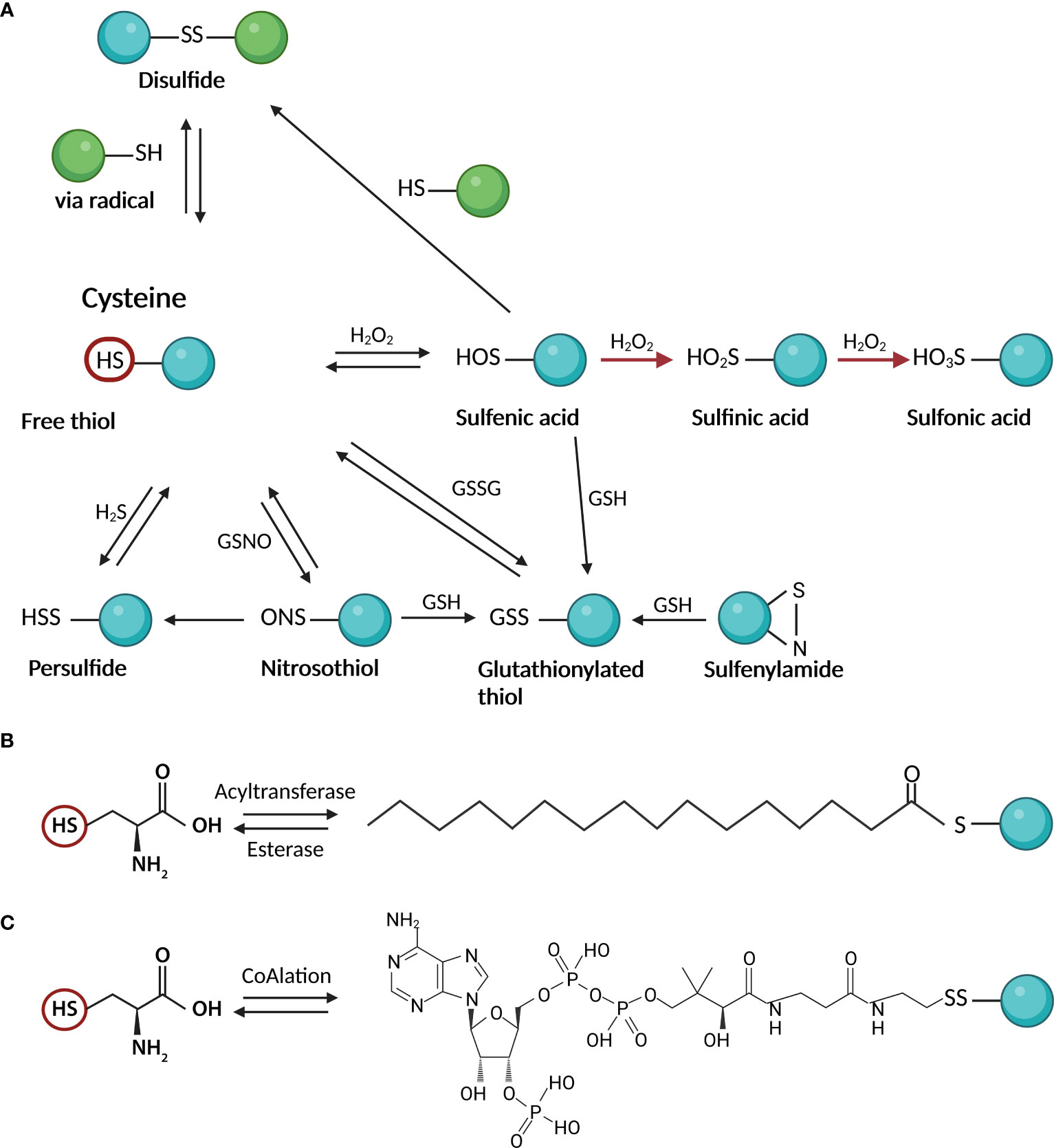
Figure 1 Cysteine posttranslational modifications; (A) oxidative modifications: Two-electron oxidation of Cys thiol(ate) groups by hydrogen peroxide, peroxynitrite, and other hydroperoxides, produces sulfenic acid. Sulfenic acid reacts with thiol groups to form disulfides or is further oxidized to irreversible sulfinic or sulfonic acids. Other products of oxidative reactions include S-nitrosothiols and persulfides; (B) S-palmitoylation as an example of S – acylation reaction: In the cells, the reaction is mediated by acyltransferases which attach the activated fatty acid-CoA to the Cys moiety. The reversibility of the reaction is ensured by esterases; (C) CoAlation: Cysteine thiols of proteins create a reversible mixed disulfide bond called protein CoAlation. This figure was created in Biorender.com (Toronto, Canada).
The reactivity of cysteine to oxidative changes is determined by the proximity of oxidants (in addition to its chemical properties). In β-cells, these are mainly superoxide (O2·−) and other oxidizing byproducts generated in mitochondria, reduced flavoprotein oxidases or monooxygenases and NADPH oxidase (NOX) family; and NO produced by NOS and combining with O2·− also peroxinitrite (16). Endogenous antioxidant system such as superoxide dismutase (SOD), peroxiredoxins (Prdx), glutathione peroxidases (GPX) and compounds such as, glutathione (GSH) and thioredoxin (Trx) are well compartmentalized (17). Together they play a critical role in maintaining redox equilibrium.
4 Other posttranslational modifications of cysteines
4.1 S-glutathionylation
S-glutathionylation is the attachment of bulky GSH to a cysteine residue via the formation of disulfides, thereby regulating protein functions in response to oxidants (18) (Figure 1A). The essential functions of reversible glutathionylation have emerged in physiology, including cardiovascular regulation (18, 19) inflammation and infection (20, 21), apoptosis (22), and cancer (23). S-glutathionylation occurs through nucleophilic sulfur chemistry in which the thiolate anion (S−) reacts with oxidized glutathione (GSSG) or reactions of GSH with electrophilic sulfur intermediates, such as sulfenic acid, S-nitrosothiol, or thiyl radical (24). The formation of glutathionylate is also balanced by the activities of various enzymes, including glutaredoxins (Grx) and glutathione transferase pi and omega (GSTP and GSTO) (25).
4.2 S-acylation
S-acylation is the covalent attachment of various fatty acids (14-20 carbon atoms) to cysteine residues via a thioester bond (Figure 1B). While most lipid modifications to proteins are irreversible, S-acylation is reversible and can be very dynamic (26). Lipid modifications can alter protein transport and membrane localization, interactions, stability, and conformation (26). While S-acylation of cysteines has mainly involved saturated palmitic acid, other studies described Cys modification by oleic acid (27–29), stearic acid (28, 30), and arachidonic acid (29). This can lead to different functional outcomes (29, 30). It depends on the exogenous supply of fatty acids reflecting cellular lipid profiles (31). The added lipid species also may vary from cysteine to cysteine on the same protein (32). The prevalence or functional significance of fatty acid type on S-acylated proteins or modified cysteine in terms of its potential for oxidative modification is not fully understood.
4.3 S-CoAlation
In recent years the antioxidant function of a key cellular metabolite, coenzyme A (CoA), has been discovered (33). CoA is an essential cofactor in all organisms and its biosynthesis involves enzymatic conjugation of cysteine, pantothenate and ATP (34). Similar to the antioxidant role of glutathione and glutathionylation of proteins, CoA protects cysteine thiols from hyperoxidation during oxidative stress by forming a mixed disulfide bond, protein CoAlation (33) (Figure 1C). Recent research on protein CoAlation has shown that it is a widespread and reversible posttranslational modification. Established cell lines and organisms have been shown to exhibit increased levels of CoAlated proteins upon oxidative or metabolic stress (33). To date, CoAlation was found to modulate the activity of modified proteins and induce significant conformational changes (35).
5 Relevance of cysteine oxidation in pancreatic β-cells physiology
For many years, β-cells were attributed as having weak antioxidant defense. However, this was based on comparison with liver or kidney, which are highly specialized detoxifying organs (36). Recently, it was shown that β-cells express Prdxs, Trxs, and SOD1/2. Thus, superoxide formed can be rapidly dismutated to H2O2, which can then serve as a signaling molecule due to its stability, thiol selectivity, and ability to diffuse through membranes. As mentioned earlier, thiol-containing molecules, Prxs/Trxs exhibit a high affinity for oxidants and are therefore efficient sensors for a prooxidant redox environment. Pancreatic β-cells are glucose sensors responding to high glucose by insulin secretion. Glucose stimulation induces oxidative metabolism, which increases the prooxidant redox status. We and others have shown that short-term glucose stimulation, which increases prooxidant status via cytoplasmic NOX4, is necessary for efficient insulin secretion, whereas long-term stimulation, which tends to induce oxidative stress, can lead to the development of T2D (37, 38). A possible mechanism for the role of prooxidant metabolism in pancreatic β-cells has been proposed (39–46).
The most important, but still technically elusive problem in redox signaling is the compartmentalization of redox status. Cellular localization then determines the source of ROS/RNS/RSS and their proximity to potential cysteine modifications, type of antioxidant defense, and the redox potential of cysteine residues present. Subcellular organelles maintain different pH values and redox potentials as well as concentrations of reactive metabolites that trigger specific redox signals (47, 48). The most important compartments for redox signaling in pancreatic β-cells are the mitochondria, the cytoplasm with the plasma membrane rafts, while oxidation in ER with the Golgi apparatus has more structural functions. Whereas the cytoplasm and mitochondria of quiescent cells have a more reduced environment (-200 and -300mV for the GSH/GSSG redox pair), which gives them a broad probability for redox signaling, e.g. upon induction of proliferation or some other stimulus, ER and Golgi secretion machinery exhibit a more oxidized potential (-150 and -140mV for the GSH/GSSG redox pair) (9, 49). This oxidized status is rather essential for the formation and maintenance of the structural disulfide bonds of the secretory proteins and counteracts redox signaling. It is difficult to determine the reduction potential of individual cellular compartments because different redox pairs are present (GSH/GSSG, Trx, Cys/Cys, etc.). They have individually different midpoint potentials that vary in different organelles. For example, Trx redox pairs are generally more reducing than GSH/GSSG, and while Trx1 shows a redox potential of -280 and -300 mV in the cytosol and nucleus, Trx2 shows an, even more, reducing redox pair of -340 and -360 mV in mitochondria (9, 48). The ability of cysteine to be modified by redox depends on many factors as stated above, but also depends on the presence of molecules with greater reactivity toward thiols such as peroxymonocarbonate. CO2 is in equilibrium with bicarbonate, which forms the biological buffer within cells. Bicarbonate can react with peroxide to form peroxymonocarbonate. The rate of its formation increases with decreasing pH (50). Thus, cellular metabolism and its compartmentalization are the main trigger for redox signaling.
5.1 Endoplasmic reticulum and Golgi secretion machinery
The importance of the ER/Golgi secretory machinery in β-cells lies in proper insulin maturation and insulin granule formation apart from lipid biosynthesis and folding, glycosylation, trafficking, and secretion of many proteins. Insulin signaling is a key function of β-cells in response to glucose. The acidic environment of ER and the progressive proton gradient from the Golgi to secretory vesicles allow the disulfide bond formation as a PTM not only in insulin molecules. Disulfide formation also mediates biomolecule degradation and ligand dissociation from receptors in the endocytic pathway. Insulin requires 3 disulfide bonds to be formed by the oxidase activity of protein disulfide isomerase (PDI). Electron transfer between PDI and ERO1A provides an oxidizing environment and disulfide bond formation (51). PDI must be reoxidized by Prdx4 in rodents and humans together with Gpx7/8 in humans, with H2O2 supplied mainly by ERO1 or the present NOX enzymes (52). The tight regulation of ERO1A activity depends on the specific formation of disulfide pairs in the protein backbone (Cys94-Cys99 for the active form vs. Cys94-131 and Cys99-104 for the inactive form) and the quality of folded proteins is thus dependent on the ER redox status (53). Disturbed ER redox homeostasis leads to ER stress, which activates the unfolded protein response. This can multiply ROS production and impair insulin production. Interestingly, Gpx7/8 show low GSH activity due to the absence of domains bound to GSH, but it plays a role in calcium storage (54). Calcium signaling has a key regulatory function for the insulin secretion machinery and functional synchronization of β-cell in the pancreatic islet. The expression of Gpx8 is regulated by Nrf2, which is involved in ER calcium management via the ATPase SERCA (55).
5.2 Insulin signaling and redox regulation in β-cells
Insulin is an important regulator of energy metabolism, affecting proliferation and survival. Its receptors are present in many tissues important for glucose uptake and utilization, such as adipose tissue, skeletal muscle, and liver. However, they are also found in the brain, kidney, heart and pancreas. A functional insulin receptor (IR) ready for insulin binding requires the formation of disulfide bonds between the receptor´s subunits, and further signal propagation involves the formation of additional disulfide bonds that regulate its activity. These proteins are kinases and phosphatases as downstream signaling cascades trigger many intracellular phosphorylation events. Insulin receptor substrate-1 (IRS1) is a signaling protein that is phosphorylated by IR and activates downstream signaling pathways. Not only does it undergo cysteine oxidation to stabilize the molecule by forming cysteine bonds, but it is regulated by S-nitrosylation, which affects downstream insulin signaling through Akt phosphorylation. Several studies suggest an important role of IRS-1 S-nitrosylation in insulin resistance through the degradation of IRS1 via the ubiquitin-proteasome pathway (56–58). The effect of S-nitrosylation on IRS1 in adipocytes is more inhibitory for glucose uptake. S-nitrosylation decreases tyrosine phosphorylation of IRS1 and activation of Akt. This effect is thought to be mediated by inhibition of protein tyrosine phosphatases (PTPs) by S-nitrosylation, resulting in increased serine phosphorylation of IRS1 and decreased tyrosine phosphorylation (59, 60). Another example of redox regulation of insulin receptors has been shown for IGFs. IGF1 and IGF2 are growth factors that share significant structural homology with insulin and bind to IR. Apart from the role that cysteine residues in IGF1 and IGF2 play in their structural stability and activity, they may form disulfide bonds with other proteins, such as IGF binding proteins (IGFBPs), which may regulate the activity and availability of these growth factors in the extracellular space (61). In any case, IR phosphorylation is regulated by the well-characterized protein tyrosine phosphatase 1B (PTBP1B) (62). Direct oxidation of PTBP1B is rather slow (~101 M-1. s-1) and it has been suggested that abundant peroxiredoxins having a higher affinity for oxidation, compete for H2O2 in cells (4, 63). The mechanism of PTBP1B oxidation occurs through the synergy of two independent mechanisms. First, bicarbonate/CO2 accelerates the oxidation of phosphatase and the same system facilitates the inhibition of peroxiredoxins by hyperoxidation, thus enabling the oxidation and inactivation of PTBP1B (63). Moreover, the activity of phosphatase is dependent on the pH in cells, and its activity increases at lower pH. This is consistent with the dependence of phosphatase oxidation on pH, which increases with higher pH (63, 64). Moreover, PTBP1B has been shown to be partially glutathionylated (at Cys215), resulting in decreased activity (65). Similarly, it has been suggested that S-nitrosylation reversibly inactivates PTBP1B, which may protect the protein from permanent inactivation by oxidative stress (66). Thus, the redox regulation of this PTBP1B is quite complex and largely depends on the metabolic state. S-glutathionylation has also been described for other downstream effectors of insulin signaling pathways, PI3K-Akt, Ras-MEKK1 (e.g. PTEN, IKKβ, NFκB, MEKK1, etc.) during the development of diabetes and some diabetic models (more in (67)), reducing their activity (Figure 2).
5.3 S-acylation in β-cells
S-acylation has been shown to be an important regulator of ion channels, vesicle trafficking, and small GTPAses, controlling many aspects of protein sorting, membrane localization, and lipid metabolism (68). S-acylated proteins of SNARE complex, including syntaxin, SNAP25, VAMP2 and synaptotagmin 1 are involved in the synaptic vesicle fusion machinery in neuronal cells (69) and also in pancreatic β-cells (70). KATP channel activity is strongly modulated by S-acylation, which causes channel opening, counteracting the ability of ATP to close the channel (71, 72). Stimulating glucose levels, however, are still able to close the channel, probably by rapid long-chain-CoA esterification to diacylglycerol (68, 73). S-acylation has also been shown to modulate the activity of voltage-gated Ca2+ channels (74) and BK channels (75), thereby affecting the electrical excitability of pancreatic β-cells by controlling action potential amplitude, depolarization, and repolarization that determine Ca2+ influx and insulin exocytosis (76) (Figure 2). Among other targets, adenine nucleotide translocase (ANT) has been shown to be inhibited by S-acylation, resulting in a decrease in the ATP : ADP ratio and increased ROS production (77).
Figure 2
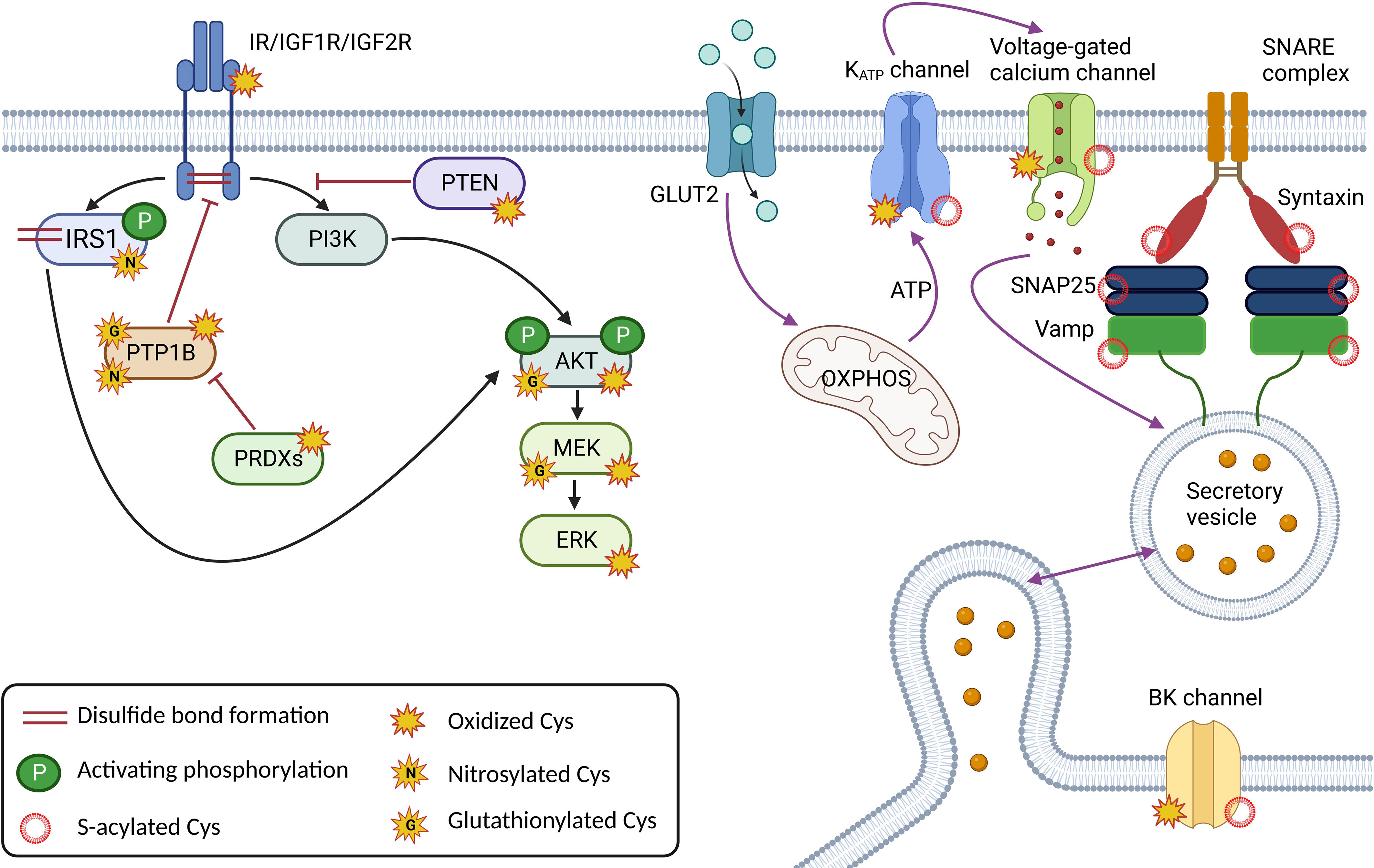
Figure 2 Major cysteine modifications in insulin and glucose signaling in β-cells; Correct redox signaling is an important requirement for healthy β-cells. Insulin receptor (IR) and Insulin-like growth factor receptors (IGF1/2R) create dimers via the disulfide bond formation, and oxidative modifications and nitrosylation are required for their activity. The redox signal is relayed by nitrosylation, glutathionylation, and/or oxidative modifications of downstream effectors such as the insulin receptor substrate (IRS1) and the kinases PI3K, Akt, MEK and ERK. The action of the kinases is regulated by the phosphatases PTP1B and PTEN, which are also subject to redox regulation. Components of the insulin secretory machinery, including ion channels at the plasma membrane such as KATP, voltage-gated calcium channels and BK channels and proteins of the SNARE complex (SNAP25, Vamp, Syntaxin), are regulated by S-acylation. The Cys of ion channels are also oxidatively modified. This figure was created in Biorender.com (Toronto, Canada).
6 Conclusion
It is obvious that not all Cys residues are the same and that the Cys residues that confer signaling properties to proteins are unique because of their position in the protein, the surrounding amino acids, protein cellular localization, and its redox environment. Although we have extensive knowledge of the location of cysteine modifications in individual proteins, we lack a functional understanding and interplay of the various cysteine modifications in a single protein and their consequences on the global scale. It is well known that β-cells require redox signaling through cysteine modifications for efficient glucose-stimulated insulin secretion and insulin signaling because of their sensitive redox homeostasis. However, metabolic overload and imbalanced redox homeostasis may lead to the development of T2D. This increases the need for research in this area as a potential intervention in the treatment of diabetes development.
Author contributions
Conceptualization: BH and LP-H; Writing the original manuscript: BH and LP-H; Manuscript review and editing: BH and LP-H; Funding acquisition: LP-H. LP-H is the guarantor of this work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Grant Agency of the Czech Republic, grant No. 22- 11439S to LP-H and by the project National Institute for Research of Metabolic and Cardiovascular Diseases (Programme EXCELES, ID Project No. LX22NPO5104) - funded by the European Union Next Generation EU.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med (2015) 80:148–57. doi: 10.1016/j.freeradbiomed.2014.11.013
2. Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol (2010) 404:902–16. doi: 10.1016/j.jmb.2010.09.027
3. Flohé L. The fairytale of the GSSG/GSH redox potential. Biochim Biophys Acta (2013) 1830:3139–42. doi: 10.1016/j.bbagen.2012.10.020
4. Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol (2008) 4:278–86. doi: 10.1038/nchembio.85
5. Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal (2013) 18:1623–41. doi: 10.1089/ars.2012.4973
6. Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radical Biol Med (2008) 45:549–61. doi: 10.1016/j.freeradbiomed.2008.05.004
7. Jensen KS, Hansen RE, Winther JR. Kinetic and thermodynamic aspects of cellular thiol-disulfide redox regulation. Antioxid Redox Signal (2009) 11:1047–58. doi: 10.1089/ars.2008.2297
8. Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol (2008) 295:C849–68. doi: 10.1152/ajpcell.00283.2008
9. Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med (2008) 44:921–37. doi: 10.1016/j.freeradbiomed.2007.11.008
10. Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res (2013) 112:382–92. doi: 10.1161/CIRCRESAHA.112.268680
11. Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, et al. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem (2013) 288:14170–7. doi: 10.1074/jbc.M113.460881
12. Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol (2004) 44:325–47. doi: 10.1146/annurev.pharmtox.44.101802.121735
13. Kabil O, Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal (2014) 20:770–82. doi: 10.1089/ars.2013.5339
14. Smith BC, Marletta MA. Mechanisms of s-nitrosothiol formation and selectivity in nitric oxide signaling. Curr Opin Chem Biol (2012) 16:498–506. doi: 10.1016/j.cbpa.2012.10.016
16. Plecitá-Hlavatá L, Ježek P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int J Biochem Cell Biol (2016) 80:31–50. doi: 10.1016/j.biocel.2016.09.010
17. Jena AB, Samal RR, Bhol NK, Duttaroy AK. Cellular red-ox system in health and disease: the latest update. Biomed pharmacother = Biomed pharmacotherapie (2023) 162:114606. doi: 10.1016/j.biopha.2023.114606
18. Rashdan NA, Shrestha B, Pattillo CB. S-glutathionylation, friend or foe in cardiovascular health and disease. Redox Biol (2020) 37:101693. doi: 10.1016/j.redox.2020.101693
19. Burns M, Rizvi SHM, Tsukahara Y, Pimentel DR, Luptak I, Hamburg NM, et al. Role of glutaredoxin-1 and glutathionylation in cardiovascular diseases. Int J Mol Sci (2020) 21:6803. doi: 10.3390/ijms21186803
20. Chia SB, Elko EA, Aboushousha R, Manuel AM, van de Wetering C, Druso JE, et al. Dysregulation of the glutaredoxin/S-glutathionylation redox axis in lung diseases. Am J Physiol Cell Physiol (2020) 318:C304–c327. doi: 10.1152/ajpcell.00410.2019
21. Mullen L, Mengozzi M, Hanschmann EM, Alberts B, Ghezzi P. How the redox state regulates immunity. Free Radic Biol Med (2020) 157:3–14. doi: 10.1016/j.freeradbiomed.2019.12.022
22. Anathy V, Roberson EC, Guala AS, Godburn KE, Budd RC, Janssen-Heininger YM. Redox-based regulation of apoptosis: s-glutathionylation as a regulatory mechanism to control cell death. Antioxid Redox Signal (2012) 16:496–505. doi: 10.1089/ars.2011.4281
23. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell (2020) 38:167–97. doi: 10.1016/j.ccell.2020.06.001
24. Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal (2011) 15:233–70. doi: 10.1089/ars.2010.3540
25. Matsui R, Ferran B, Oh A, Croteau D, Shao D, Han J, et al. Redox regulation via glutaredoxin-1 and protein s-glutathionylation. Antioxid Redox Signal (2020) 32:677–700. doi: 10.1089/ars.2019.7963
26. Chen JJ, Fan Y, Boehning D. Regulation of dynamic protein s-acylation. Front Mol Biosci (2021) 8:656440. doi: 10.3389/fmolb.2021.656440
27. Montigny C, Decottignies P, Le Maréchal P, Capy P, Bublitz M, Olesen C, et al. S-palmitoylation and s-oleoylation of rabbit and pig sarcolipin. J Biol Chem (2014) 289:33850–61. doi: 10.1074/jbc.M114.590307
28. Thinon E, Percher A, Hang HC. Bioorthogonal chemical reporters for monitoring unsaturated fatty-acylated proteins. Chembiochem Eur J Chem Biol (2016) 17:1800–3. doi: 10.1002/cbic.201600213
29. Liang X, Nazarian A, Erdjument-Bromage H, Bornmann W, Tempst P, Resh MD. Heterogeneous fatty acylation of src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J Biol Chem (2001) 276:30987–94. doi: 10.1074/jbc.M104018200
30. Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, Miller AK, et al. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature (2015) 525:124–8. doi: 10.1038/nature14601
31. Muszbek L, Haramura G, Cluette-Brown JE, Van Cott EM, Laposata M. The pool of fatty acids covalently bound to platelet proteins by thioester linkages can be altered by exogenously supplied fatty acids. Lipids (1999) 34 Suppl:S331–7. doi: 10.1007/BF02562334
32. Brett K, Kordyukova LV, Serebryakova MV, Mintaev RR, Alexeevski AV, Veit M. Site-specific s-acylation of influenza virus hemagglutinin: the location of the acylation site relative to the membrane border is the decisive factor for attachment of stearate. J Biol Chem (2014) 289:34978–89. doi: 10.1074/jbc.M114.586180
33. Tsuchiya Y, Peak-Chew SY, Newell C, Miller-Aidoo S, Mangal S, Zhyvoloup A, et al. Protein CoAlation: a redox-regulated protein modification by coenzyme a in mammalian cells. Biochem J (2017) 474:2489–508. doi: 10.1042/BCJ20170129
34. Leonardi R, Zhang YM, Rock CO, Jackowski S, Coenzyme A. Back in action. Prog Lipid Res (2005) 44:125–53. doi: 10.1016/j.plipres.2005.04.001
35. Tossounian MA, Baczynska M, Dalton W, Newell C, Ma Y, Das S, et al. Profiling the site of protein CoAlation and coenzyme a stabilization interactions. Antioxidants (Basel Switzerland) (2022) 11:1362. doi: 10.3390/antiox11071362
36. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radical Biol Med (1996) 20:463–6. doi: 10.1016/0891-5849(96)02051-5
37. Plecita-Hlavata L, Jaburek M, Holendova B, Tauber J, Pavluch V, Berkova Z, et al. Glucose-stimulated insulin secretion fundamentally requires H2O2 signaling by NADPH oxidase 4. Diabetes (2020) 69:1341–54. doi: 10.2337/db19-1130
38. Benáková Š., Holendová B, Plecitá-Hlavatá L. Redox homeostasis in pancreatic β-cells: from development to failure. Antioxidants (Basel Switzerland) (2021) 10. doi: 10.3390/antiox10040526
39. Mittal M, Gu XQ, Pak O, Pamenter ME, Haag D, Fuchs DB, et al. Hypoxia induces kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med (2012) 52:1033–42. doi: 10.1016/j.freeradbiomed.2011.12.004
40. Ferdaoussi M, MacDonald PE. Toward connecting metabolism to the exocytotic site. Trends Cell Biol (2017) 27:163–71. doi: 10.1016/j.tcb.2016.10.003
41. Lorenzen I, Eble JA, Hanschmann EM. Thiol switches in membrane proteins - extracellular redox regulation in cell biology. Biol Chem (2020) 402:253–269. doi: 10.1515/hsz-2020-0266
42. Corkey BE, Shirihai O. Metabolic master regulators: sharing information among multiple systems. Trends Endocrinol metabolism: TEM (2012) 23:594–601. doi: 10.1016/j.tem.2012.07.006
43. Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocrine Rev (2008) 29:317–33. doi: 10.1210/er.2007-0039
44. Oslowski CM, Hara T, O'Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab (2012) 16:265–73. doi: 10.1016/j.cmet.2012.07.005
45. Sokolova M, Sahraoui A, Høyem M, Øgaard J, Lien E, Aukrust P, et al. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am J Physiol Endocrinol Metab (2018) 315:E912–e923. doi: 10.1152/ajpendo.00461.2017
46. Xu Z, Lam LS, Lam LH, Chau SF, Ng TB, Au SW. Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J (2008) 22:127–37. doi: 10.1096/fj.06-7871com
47. Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol (2010) 11:50–61. doi: 10.1038/nrm2820
48. Jones DP, Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab (2010) 12 Suppl 2:116–25. doi: 10.1111/j.1463-1326.2010.01266.x
49. Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med (1999) 27:1208–18. doi: 10.1016/S0891-5849(99)00145-8
50. Bakhmutova-Albert EV, Yao H, Denevan DE, Richardson DE. Kinetics and mechanism of peroxymonocarbonate formation. Inorg Chem (2010) 49:11287–96. doi: 10.1021/ic1007389
51. Benham AM, van Lith M, Sitia R, Braakman I. Ero1-PDI interactions, the response to redox flux and the implications for disulfide bond formation in the mammalian endoplasmic reticulum. Philos Trans R Soc Lond B Biol Sci (2013) 368:20110403. doi: 10.1098/rstb.2011.0403
52. Zito E, Melo EP, Yang Y, Wahlander Å., Neubert TA, Ron D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol Cell (2010) 40:787–97. doi: 10.1016/j.molcel.2010.11.010
53. Inaba K, Masui S, Iida H, Vavassori S, Sitia R, Suzuki M. Crystal structures of human Ero1α reveal the mechanisms of regulated and targeted oxidation of PDI. EMBO J (2010) 29:3330–43. doi: 10.1038/emboj.2010.222
54. Yoboue ED, Rimessi A, Anelli T, Pinton P, Sitia R. Regulation of calcium fluxes by GPX8, a type-II transmembrane peroxidase enriched at the mitochondria-associated endoplasmic reticulum membrane. Antioxid Redox Signal (2017) 27:583–95. doi: 10.1089/ars.2016.6866
55. Granatiero V, Konrad C, Bredvik K, Manfredi G, Kawamata H. Nrf2 signaling links ER oxidative protein folding and calcium homeostasis in health and disease. Life Sci alliance (2019) 2:e201900563. doi: 10.26508/lsa.201900563
56. Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, et al. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes (2005) 54:959–67. doi: 10.2337/diabetes.54.4.959
57. Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, et al. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem (2005) 280:14203–11. doi: 10.1074/jbc.M411226200
58. Pilon G, Charbonneau A, White PJ, Dallaire P, Perreault M, Kapur S, et al. Endotoxin mediated-iNOS induction causes insulin resistance via ONOO- induced tyrosine nitration of IRS-1 in skeletal muscle. PloS One (2010) 5:e15912. doi: 10.1371/journal.pone.0015912
59. Wang H, Wang AX, Aylor K, Barrett EJ. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes (2013) 62:4030–42. doi: 10.2337/db13-0627
60. Ropelle ER, Pauli JR, Cintra DE, da Silva AS, De Souza CT, Guadagnini D, et al. Targeted disruption of inducible nitric oxide synthase protects against aging, s-nitrosation, and insulin resistance in muscle of male mice. Diabetes (2013) 62:466–70. doi: 10.2337/db12-0339
61. Sitar T, Popowicz GM, Siwanowicz I, Huber R, Holak TA. Structural basis for the inhibition of insulin-like growth factors by insulin-like growth factor-binding proteins. Proc Natl Acad Sci U.S.A. (2006) 103:13028–33. doi: 10.1073/pnas.0605652103
62. Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes (2005) 54:311–21. doi: 10.2337/diabetes.54.2.311
63. Netto LES, Machado L. Preferential redox regulation of cysteine-based protein tyrosine phosphatases: structural and biochemical diversity. FEBS J (2022) 289:5480–504. doi: 10.1111/febs.16466
64. Groen A, Lemeer S, van der Wijk T, Overvoorde J, Heck AJ, Ostman A, et al. Differential oxidation of protein-tyrosine phosphatases. J Biol Chem (2005) 280:10298–304. doi: 10.1074/jbc.M412424200
65. Rinna A, Torres M, Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med (2006) 41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010
66. Chen YY, Chu HM, Pan KT, Teng CH, Wang DL, Wang AH, et al. Cysteine s-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem (2008) 283:35265–72. doi: 10.1074/jbc.M805287200
67. Sánchez-Gómez FJ, Espinosa-Díez C, Dubey M, Dikshit M, Lamas S. S-glutathionylation: relevance in diabetes and potential role as a biomarker. Biol Chem (2013) 394:1263–80. doi: 10.1515/hsz-2013-0150
68. Corkey BE, Deeney JT, Merrins MJ. What regulates basal insulin secretion and causes hyperinsulinemia? Diabetes (2021) 70:2174–82. doi: 10.2337/dbi21-0009
69. Prescott GR, Gorleku OA, Greaves J, Chamberlain LH. Palmitoylation of the synaptic vesicle fusion machinery. J neurochem (2009) 110:1135–49. doi: 10.1111/j.1471-4159.2009.06205.x
70. Cheng H, Straub SG, Sharp GW. Protein acylation in the inhibition of insulin secretion by norepinephrine, somatostatin, galanin, and PGE2. Am J Physiol Endocrinol Metab (2003) 285:E287–94. doi: 10.1152/ajpendo.00535.2002
71. Larsson O, Deeney JT, Branstrom R, Berggren PO, Corkey BE. Activation of the ATP-sensitive k+ channel by long chain acyl-CoA. a role in modulation of pancreatic beta-cell glucose sensitivity. J Biol Chem (1996) 271:10623–6. doi: 10.1074/jbc.271.18.10623
72. Tarasov A, Dusonchet J, Ashcroft F. Metabolic regulation of the pancreatic beta-cell ATP-sensitive k+ channel: a pas de deux. Diabetes (2004) 53 Suppl 3:S113–22. doi: 10.2337/diabetes.53.suppl_3.S113
73. Lorenz MA, El Azzouny MA, Kennedy RT, Burant CF. Metabolome response to glucose in the β-cell line INS-1 832/13. J Biol Chem (2013) 288:10923–35. doi: 10.1074/jbc.M112.414961
74. Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L. Unique regulatory properties of the type 2a Ca2+ channel beta subunit caused by palmitoylation. Proc Natl Acad Sci U.S.A. (1998) 95:4690–5. doi: 10.1073/pnas.95.8.4690
75. Duncan PJ, Bi D, McClafferty H, Chen L, Tian L, Shipston MJ. S-acylation controls functional coupling of BK channel pore-forming alpha-subunits and beta1-subunits. J Biol Chem (2019) 294:12066–76. doi: 10.1074/jbc.RA119.009065
76. Chamberlain LH, Shipston MJ, Gould GW. Regulatory effects of protein s-acylation on insulin secretion and insulin action. Open Biol (2021) 11:210017. doi: 10.1098/rsob.210017
77. Ciapaite J, Bakker SJ, Diamant M, van Eikenhorst G, Heine RJ, Westerhoff HV, et al. Metabolic control of mitochondrial properties by adenine nucleotide translocator determines palmitoyl-CoA effects. Implications Mech linking Obes type 2 diabetes FEBS J (2006) 273:5288–302. doi: 10.1111/j.1742-4658.2006.05523.x
Keywords: cysteine, thiol, pancreatic beta cells, posttranslational modifications, redox signaling
Citation: Holendova B and Plecita-Hlavata L (2023) Cysteine residues in signal transduction and its relevance in pancreatic beta cells. Front. Endocrinol. 14:1221520. doi: 10.3389/fendo.2023.1221520
Received: 12 May 2023; Accepted: 13 June 2023;
Published: 29 June 2023.
Edited by:
Elena Silvestri, University of Sannio, ItalyReviewed by:
Leonor Thomson, University of the Republic, UruguayCopyright © 2023 Holendova and Plecita-Hlavata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lydie Plecita-Hlavata, lydie.plecita@fgu.cas.cz