
Abstract
In the present work, ring-shaped samples made from steel 20MnCr5 were low-pressure carburized (LPC) and subsequently hardened by gas quenching (case-hardened). This results in a near-surface gradient in chemical composition, microstructure- and hardness distribution, as well as a three-dimensional residual stress (RS) distribution, which was investigated by neutron diffraction. Near-surface RSs in the ferrite-/martensite- and austenite phase are additionally determined by X-ray diffraction. It is shown that the chemical gradient has an influence on the chosen \({d}_{0}\) strategy and how such a reference sample should be extracted. If near-surface RS values are to be determined by neutron diffraction, the pseudo-strain effect must be taken into account. For this purpose, a suitable approach using the “open source” software SIMRES and STRESSFIT is also presented. By combining neutron and X-ray diffraction data, a complete RS distribution over the whole sample can be obtained.
Similar content being viewed by others


1 Introduction
The properties of the outer surface layers are of utmost importance for the functional capability and lifetime of highly loaded structural and functional components. Many failure processes, such as the initiation of fatigue cracks, originate at the very surface. For steel components this hardening is mainly realized by means of hardening processes (i.e., References 1,2,3,4). For hardenable steels, the most common approach is the application of heat treatment processes aimed at forcing the microstructural transformation to martensite via rapid quenching from austenitizing temperature. The maximum achievable hardness is determined in particular by the dissolved carbon content. In addition, this also significantly determines the transformation kinetics during quenching. If a high basic toughness of the component is to be combined with a high hardness in the near-surface layers, a common approach is to use a case-hardening steel with a relatively low nominal carbon content (typically in the range of 0.15 to 0.25 Ma pct) and to carry out a carburizing before quenching. By this means, through the inward diffusion of carbon into the near-surface volume and the subsequent quenching a hard layer is generated. However, this case-hardening process always creates a chemical gradient, i.e. the carbon content decreases with increasing depth, and at the same time diffusion and effusion processes can also affect the local distribution of other chemical elements of the applied steel grade.[5,6]
Case-hardening always results in the generation of characteristic residual stress (RS) depth distributions. Generally, compressive RS are induced near the surface within the hardened layer and balancing tensile RS are created in the core. In most cases, these near-surface compressive RS introduced by hardening and tempering can impede crack initiation and propagation and are therefore considered beneficial for engineering applications. The evaluation of process-induced RS is crucial for the reliable assessment of the quality and service life of a component.
Various methods and techniques exist for analysing RS depth distributions. In general, these can be divided into mechanical, acoustic, magnetic and diffraction methods. The most frequently used methods in science and industrial practice are the mechanical methods, especially the incremental hole drilling method[7] and the group of the diffraction methods. However, all mechanical methods have in common that they are at least partially destructive as they are based on interfering the RS equilibrium by inserting a mechanical separation cut (as e.g., drilling, milling). In contrast, diffraction methods can be used to non-destructively analyse RS. Here, the penetration depth of the radiation used is decisive. Low-energy X-rays have a penetration depth of only a few µm in most metals.[8,9] Therefore, a complete RS depth distribution on a workpiece can only be determined in combination with a layer removal for instance by means of electrochemical polishing and reapplication of the method at the newly generated surface. This finally also results in the destruction of the workpiece and in particular, in redistributions of the RS states between the layer removal steps, which must be corrected accordingly, especially when large information depths are required. The appropriate correction of these effects needs pre-knowledge about the triaxial RS state, which is often not available. This problem can be avoided using high energy X-rays, typically provided by synchrotron sources or using neutron radiation. In the latter case, the penetration depth is in the range of several centimetres in steels.[10,11] By this means, the entire triaxial RS state in the bulk of industrial workpieces can be determined non-destructively. When RS depth distributions close to the surface are of particular interest, through surface strain scanning approaches have been proposed. These approaches have the disadvantage that during scanning the nominal gauge volume, which is defined by optics used in the primary and secondary neutron beam paths (slit systems, radial collimators, parabolic guides) of the neutron diffraction set-up, through the surface, this nominal gauge volume is only partially filled by material. A nominal gauge volume only partially filled with material will cause a so-called “surface effect”,[10,12,13] which generates shifts of the recorded peak positions, which are not related to strain.[12] These shifts can lie in a range that corresponds to several hundred up to a few thousand microstrain. The instrumental configuration, scattering geometry and measuring strategy have most influence on this error. At first, there is a geometric error. It appears when position sensitive detectors are used. In this case, a shift of the center of the scattering volume (sampled gauge volume) is projected to the detector and appears as a peak-shift. Its contribution to pseudo-strain is several hundred microstrain and depends on the size of the instrumental gauge volume and the detector distance. The second contribution to pseudo-strain is the wavelength distribution in the primary beam. It is an effect of focusing monochromators, generated to refocus the diffracted beam, thus improving strain resolution, while using a maximum beam divergence in order to gain intensity. Another minor effect is the absorption in the sample material, which shifts the center of gravity of the diffracting volume within the sampled gauge volume.
For the present experiment on SALSA we used radial focusing collimators as beam shaping optics, which reduce the pseudo-strain significantly[14,15] while accepting maximum beam divergence and providing a precisely defined gauge volume. In addition, we performed vertical scans to minimize the associated error, because the above-described surface effect appears in the scattering plane only—which is horizontally on SALSA. The correction of pseudo-strain is performed by a reference measurement to determine instrumental parameters, which are then used within a computer model for pseudo-strain correction.
This must be taken into account when dealing with RS depth distributions induced by the process of case-hardening, which typically induces gradients in the chemical composition and in the RS distributions in the depth range of a few tenths of a millimetre up to some millimetres depending on the process parameters applied. To determine RS depth gradients by means of neutron diffraction stress analysis lattice strains must be determined in depths close to the surface using a relative large nominal gauge volume, which is typically in the range of 1 × 1 × 1 mm3 and even larger.
For correct RS evaluation, the occurring spurious strains must be taken into account and either minimized by a suitable measurement strategy or corrected by suitable simulations of the diffraction experiment. For single-phase and gradient-free materials, in previous work, this correction was successfully performed using the Monte Carlo simulation software “SIMRES”[16,17,18] and “STRESSFIT”,[19,20] respectively. It was also shown in Reference 21 that a correction with “SIMRES” is also applicable in the presence of near-surface RS gradients in coarse multiphase materials. To which extent this approach can be applied to materials where a further gradient in the local chemical composition occurs will be part of the current work.
A further question that generally arises for the application of neutronographic RS analysis for case-hardened steels is on the appropriate provision of the reference value \({d}_{0}\), which is the interplanar spacing for the unstressed crystal lattice. This reference value is essential for proper calculation of lattice strains. With regard to a suitable \({d}_{0}\) strategy, there are various approaches as to how an appropriate reference value can be determined. Powder specimens, free-cut reference samples or the measurement of sample areas that can be assumed to be stress-free have proven to be successful.[22] Alternatively, a suitable constant value for the strain-free lattice parameter can be calculated by assuming stress equilibrium.[22,23] In the case of components that exhibit a microstructural or chemical gradient, respectively, the methods mentioned above, with the exception of the free-cut reference samples, are not useful, since the chemical gradient directly affects the value of the strain- and stress-free lattice plane parameter \({d}_{0}\), so it is no longer constant over the whole sample, but depends on the position.[24,25] The study on the correct \({d}_{0}\) assumption for a meaningful data treatment for case-hardened material states is also the focus of the present work.
In the present paper, these questions are addressed systematically. In a first step, the existing (chemical) gradients in a carburized and case-hardened ring like geometry, as a model for a gear wheel, were determined precisely. In a further step, the evolution of lattice plane spacing in the three principal directions were determined neutronographically. This lattice plane spacing distribution was evaluated taking into account several \({d}_{0}\) approaches and converted into strain or stress distributions. In particular, the appropriate size of the \({d}_{0}\) reference samples was considered. In addition, it will be examined to what extent it is possible to predict a \({d}_{0}\) distribution based on the initial state under consideration of the chemical gradients. The near-surface RS values are to be validated with comparison with X-ray diffraction analyses. The question of a suitable \({d}_{0}\) strategy is completed by a detailed consideration of the sampling strategy of the \({d}_{0}\) reference samples. Here, the \({d}_{0}\) approach with free-cut reference samples always involves the question of the extent, to which this sampling itself contributes to influencing the near-surface material zone.
2 Materials and Methods
2.1 Sample Shape and Material Characterization
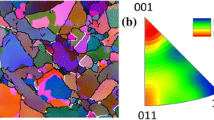
To tackle these problems a case-hardening steel 20MnCr5 (material no. 1.7147) is chosen as model material. Near-net-shape model samples were produced, i.e., rings with an outer diameter of 33 mm and an inner diameter of 15 mm and a height of 15 mm were manufactured. The sample geometry with the underlying coordinate system is shown in Figures 1(b) and (c), respectively. The samples were carburized and hardened by means of low-pressure carburizing (LPC) following an industrial processing route. The targeted values were the nominal carbon content at the surface of about 0.8 mass pct and a case-hardening depth (CHD) defined at hardness 550 HV of about 1.5 mm. As LPC process parameters, a carburizing temperature of 980 °C was chosen in combination with eight carburizing pulses. The quenching was performed with 3 bar of nitrogen gas. The samples were then single-hardened at austenitizing temperature of 880 °C and quenched with a 15 bar nitrogen gas flow. The resulting microstructure is shown in Figure 2. Spectroscopy measurements were performed on one sample to determine the chemical gradients. Glow discharge optical emission spectroscopy (GDOES) and spark optical emission spectrometry (SOES) have been applied to analyse the concentration depth profile of manganese (GDOES) and carbon (SOES), respectively.[26,27] It should be noted that a depth profile was recorded only on the lateral mantle surface and transferred to the inner and outer mantle surface. In addition, retained austenite analyses were performed on both the outer and inner mantle surfaces using an X-ray diffractometer with Zr-filtered Mo Kα radiation (wavelength 0.71 Å). A 1 × 1 mm2 collimator was used as primary aperture, while a 2 mm slit aperture was placed in the secondary beam path in front of a point detector. The 2Θ scanning range was between 20 and 58 deg with a step size of 0.1 deg. The obtained data were analyzed with the 6-line-method[28] to calculate the phase fraction of the retained austenite phase. The radiographic constants required for data evaluation (e.g., R-factors) were taken from Reference 28. A stepwise electrochemical material removal was carried out for determination of the retained austenite depth distribution.

Experimental setup at SALSA@Ill[33] (a), sample geometry (b) and coordinate system (c). The right side (b and c) shows the sample geometry including the coordinate system. OD indicates the measuring point at the outer surface, ID at the inner surface, respectively. MD indicates the measuring direction. On the left side (a) the used setup at the SALSA experiment is shown with the radial collimators used in the primary and on the secondary beam paths, respectively. In the front part of the partial image (a), the different reference samples can be seen, which were also measured in hoop direction within the setup. OD, outer diameter; ID, inner diameter

Micrographs of the cross-section of the ring-shaped case-hardened 20MnCr5 sample. The nominal CHD value was 1.5 mm. A section of the inner and outer surface of the sample is shown. The metallographically prepared sample was etched according to Klemm-I[34]
In addition, micro-hardness measurements were performed on a metallographically prepared cross-section sample. These measurements were performed using instrumented hardness testing according to ISO 14577[29,30,31,32] with a indentation testing system type Fischerscope H100 from Fischer (Sindelfingen, Germany) using a test load of 100 mN and a Vickers pyramid as indenter.
2.2 Residual Stress Analysis
Neutronographic RS analyses[33] were performed at the neutron diffraction instrument SALSA[35] at the European Research Reactor of the Institute Laue-Langevin (ILL) in Grenoble/France. The general setup is shown in Figure 1(a). The used wavelength was 1.704 Å. In the ferrite phase, the {211} lattice planes were investigated. No neutronographic measurements were performed in the austenite phase, since higher amount of retained austenite are localized to a very shallow information depth of only a few hundred micrometres, which is not easily assessable for neutron diffraction stress analysis. In the case of near-surface measurements, through surface strain scans are often applied, where the stationary nominal gauge volume may not be completely filled with sampled material. As a result, actual neutron sampling distribution is non-uniform within the gauge volume, which may lead to systematic errors of strain measurements, commonly called pseudo-strains. The stepwise sample movement for realizing this through surface scanning can be either vertical height (i.e., normal to the diffraction plane and the scattering vector) or horizontal (i.e., in a direction parallel to the diffraction plane). In the case of vertical movement, this is often termed the z-mode. The depth profile of the lattice plane spacings was determined for the hoop direction (transmission configuration), as well as the axial direction (reflection configuration) in z-mode, which is illustrated in Figures 3(a) and (b). The main advantage of the z-mode is the central symmetry of sampling in the diffraction plane, which makes the pseudo-strain effects negligible. Only the difference between the positions of the sampling centre of gravity and the nominal gauge centre in z-direction has to be considered in data analysis. If, on the other hand, the nominal gauge volume is filled from the side (horizontal sample motion, Figure 3(c), the pseudo-strains become significant due to the shift of the sampling centre of gravity and uneven wavelength distribution. The latter is described in literature as the “wavelength effect”.[10] In addition, a “positional discrimination effect” can occur in this configuration, since the position of the detectors is aligned with the nominal centre of the gauge volume.[10] In this work, the undesirable pseudo-strains were numerically corrected using the simulation software SIMRES[16,17,18] and STRESSFIT.[19,20] The ray-tracing simulation of the instrument in given experimental geometry by SIMRES provided the nominal sampling distribution (a cloud of possible scattering events). This event cloud was then used to perform the pseudo-strain correction in STRESSFIT, which performs a convolution of the nominal sampling distribution with the sample geometry. The neutronographic measurements were performed with a nominal gauge volume of 0.6 × 0.6 × 2 mm3 (x–y–z, Figure 3(d)) defined by radial collimators. On the primary side, two radial collimators with FWHM (full widths at half maximum) of 2 and 0.6 mm in vertical and horizontal direction, respectively, were used. On the secondary side, a radial collimator with FWHM of 0.6 mm was used. The diffraction lines were fitted with the evaluation software LAMP[36] using a Gaussian function as the model peak profile. As is common practice, the fit error was included in the calculation of the lattice plane spacings and subsequently also to strains or stresses by error propagation. Thus, an error estimate can be made for each measurement point.

Neutronographic measurement strategy to determine the interference line position in the three independent directions. To determine the results in the hoop (a) and axial direction (b), the sample was moved through the nominal gauge volume stepwise in z-direction. To measure the interference line position in radial direction (c), the sample was moved stepwise in x-direction. In the figure, the movement of the sample relative to the stationary measuring gauge volume is shown by red arrows. In (d) the three-dimensional sample is shown with indication of the measuring directions. The position of the neutronographic measurement and the resulting direction of movement of the sample are shown again in red. The scattering vector Q is drawn in blue (Color figure online)
The calculation of the respective lattice strain was carried out according to Reference 37:
Here, \({d}^{hkl}\) describes the experimentally determined interplanar spacing and \({d}_{0}^{hkl}\) the reference value for the strain-free state (or for the unstressed lattice). The stress was calculated according to Reference 9 using
Here \({\varepsilon }_{\mathrm{direction}}^{hkl}\) corresponds to the calculated lattice strain in one of the three examined directions (axial, hoop or radial).\({E}^{hkl}\) corresponds to the lattice plane-specific Young's modulus and \({\upsilon }^{hkl}\) to the Poissons ratio, respectively. In the context of this work, values of \({E}^{\{211\}}\) = 219,911 MPa and \({\upsilon }^{\{211\}}\) = 0.285 were applied.[38]
2.3 \({{\varvec{d}}}_{0}\)-Determination
As part of the investigation on the identification of a suitable \({d}_{0}\) strategy, two pins with diameters of Ø 1.7 and 3 mm were sectioned from an identical twin sample. The pin extraction was done centrally in the bulk sample with the longitudinal pin axis oriented parallel to a radial axis from the inner to the outer surface. The two reference pins were manufactured by means of wire electro discharge machining (wire EDM). The justification for using EDM for this purpose was given by pre-studies, which are also explained in this work. For each of these, an interplanar spacing vs. depth distribution was determined in radial direction. Here, the pin was oriented such that the measured radial direction corresponded to the hoop direction of the bulk sample. The measuring volume used was 0.6 × 0.6 × 0.6 mm3 with the same experimental setup as used for the ring samples.
In a pre-study it was investigated, which method is best suited for the sectioning of \({d}_{0}\)-reference samples. This test series was done with the quenched and tempered steel 42CrMo4 (SAE4140) as model material, which is not much different from the case-hardening steel grade used in the neutron diffraction studies. RS depth distributions induced by waterjet cutting, wire erosion machining and wire saw cutting (using standard processing parameters) were determined and compared. Stress-free annealed plates of 42CrMo4 with dimensions 40 × 20 × 10 mm3 were provided for this purpose. The cutting was done using a wire saw type 6234 from well (Mannheim Germany), a waterjet cutting system type iCUTwater SMART (imes-icore, Eiterfeld, Germany) and a wire EDM machine type BA24 (MITSUBISHI), respectively. RS depth distributions were determined using X-ray diffraction RS analysis in combination with successive electrochemical layer removal.
Further X-ray RS analysis were carried out on the case-hardened 20MnCr5 ring at the outer ring surface that was accessible without sectioning of the ring complementary to neutronographic stress analyses. All X-ray RS analyses, i.e., on the ring and for the pre-studies on the 42CrMo4 steel plates were performed according to the well-known sin2ψ method.[37] Vanadium-filtered Cr Kα- radiation was used and a 3-circle diffractometer in ψ-geometry was employed to analyse the {211} lattice planes of the ferrite/martensite phase. For all measurements, a pinhole collimator with a nominal diameter of Ø 2 mm was used as primary aperture. On the secondary side of the beam path, a 0.8 deg symmetrisation aperture was installed in front of the scintillation counter. Using X-ray diffraction also the austenite {220} lattice planes were studied for the case-hardened ring of steel 20MnCr5 in case that the amount of retained austenite phase was above 10 vol pct. Here, a 2 mm slit was applied on the secondary side. A total of 21 sample tilts was used equidistantly distributed in sin2ψ in the range between + 60 ≤ ψ ≤ − 60 deg. After subtraction of the background, the interference lines were fitted using Pearson VII functions. The kα2-correction in the austenite phase was performed using a double peak fit. The successive layer removal was carried out by means of stepwise electrochemical polishing.
3 Results and Discussion
3.1 Chemical Analysis, Microhardness, Retained Austenite
The results of the chemical analysis, i.e., the carbon and manganese content depth distributions, are shown in Figure 4. Based on these measurements, it was shown that no significant manganese effusion occurred in the surface region. The carbon content in the near-surface layer was about 0.85 mass pct showing a significant gradient in depth. In a depth of approx. 2 mm, the carbon content level of about 0.2 mass pct of the base material (20MnCr5) is reached. The quenching process during hardening results in the formation of martensite in the case (see Figure 2). In addition, a significant fraction of retained austenite phase is remaining close to the surface (see Figure 4) due to the high carbon content and the high quenching rate.

Depth distributions of the manganese concentration (GDOES, red points), the carbon concentration (SOES, black points), retained austenite (blue points) and of the Martens micro-hardness HM (orange points) over the wall thickness of the rings. OD, outer diameter; ID, inner diameter (Color figure online)
At the outer surface, the determined retained austenite phase content is about 23 ± 1 pct, and the maximum value of about 33 ± 2 pct occurs at a distance of approx. 0.06 mm from the outer surface. At the inner surface (ID), the value is approx. 25 ± 2 pct and the maximum value for retained austenite phase content of about 35 ± 2 pct occurs at a distance of approx. 0.025 mm from the inner surface, respectively. In both cases, the retained austenite phase content strongly decreases starting at a depth of approx. 0.3 mm. The resulting micro-hardness depth correlates well with increasing martensite content in the first 0.5 mm from both treated surfaces. The slight decrease in micro-hardness at both surfaces (ID and OD) is due to the high amount of retained austenite phase in this region. At larger depths, first an elevated micro-hardness plateau can be observed. Starting from a distance to the surface of about 1 mm, there is also a clear decrease in micro-hardness on both sides of the ring. Thus, the proportion of formed martensite and retained austenite seems to be more decisive for the hardness distribution than the pure carbon content. According to Reference 39, the micro-hardness distribution is influenced by the carbon depth gradient, the depth distributions of the retained austenite content as well as the macro and micro RS distributions. In Reference 39 the significant part of the resulting micro-hardness distribution is assigned to the carbon gradient, which in turn determines the depth distribution of the retained austenite content and the local phase-specific micro RSs. Starting from a depth of about 2 mm, no further change in micro-hardness is evident. According to EN ISO 2639:2002,[40] the hardness profile for a CHD 1.5 sample should correspond to a hardness value of 550 HV1 at a depth of about 1.5 mm. However, the Vickers hardness in this region derived from the Martens hardness test (HM 0.01) is somewhat higher here, indicating a slightly higher CHD as aimed for.
3.2 Reference Samples, Reference Value \({d}_{0}\)
With regard to the preparation of suitable \({d}_{0}\) reference samples, care must be taken to ensure an appropriate and sufficiently gentle sample sectioning. The results of the preliminary investigations on stress relieved annealed 42CrMo4 steel plates are presented in Figures 5(a) and (b). The results indicate that sampling by means of wire sawing has the smallest influence on the resulting RS state. As shown in Figure 5(a), by means of wire sawing compressive RS in the range between − 300 and − 500 MPa were induced, which, however, decrease rapidly with increasing distance to the surface. In the case of samples produced by waterjet cutting, comparable RS in the range of − 400 MPa occur near the surface, with an additional RS maximum of up to − 550 MPa forming below the surface. The largest RS amounts are induced during wire EDM. Here, tensile RS of up to 500 MPa are generated at the cut surface, which increase to approx. 700 MPa at a depth of about 20 µm. For all three methods, however, it can be stated that starting from a depth of about 70 to 80 µm, RS caused by the cutting process are no longer present. Regarding the average integral width of the X-ray diffraction interference lines (Figure 5(b)), these results support this finding that the sample is not affected by any of the three methods for regions beyond 70 µm depth. The significantly higher integral width with EDM presumably results from a martensitic transformation due to the induced heat. Since reference samples generally have to be very small, the issue of manageability is of particular importance. With a manual wire saw, a cylindrical sample geometry is hardly to be realized. Likewise, the waterjet cutting and electro discharge machining process can only be used for cylindrical sample (easy to adjust, uniform stress release in all directions of the x–y plane) manufacturing if the reference samples can be reliably retrieved afterwards. Likewise, the dimensional accuracy of the small reference samples cannot be guaranteed during waterjet cutting due to the involved high forces. That is, the samples will always become slightly tapered. The reason for this is the always slightly conical shape of the water jet, which prevents the formation of an ideal vertical cut surface. Hence, since RS analyses indicate that for all sectioning methods studied here in amount comparable RS distributions were induced, while the affected region is in all cases limited to about 70 µm, the wire EDM was employed for manufacturing of the present \({d}_{0}\) reference samples. The zone affected by EDM can either be removed afterwards, e.g., by electrochemical polishing, or the nominal sample volume must be smaller than the sample dimension. Based on current results, it can be recommended to section \({d}_{0}\) reference pins with a diameter that is approx. 200 to 300 µm larger than the diagonal of the gauge volume defined by the apertures in the neutron diffraction set-up. Here, an increase in diameter of 200 to 300 µm is to be understood as a conservative estimate and recommendation by the authors. This way, the reference data are not influenced by the machining induced near-surface RS gradients, assuming that accurate adjustment of the sample is feasible.

Residual stress depth distribution (a) and depth distribution of the integral widths of the X-ray diffraction lines (b) after sample sectioning experiments on stress-free annealed 42CrMo4 steel plates (40 × 20 × 10 mm3). Here TD denotes the transverse direction and ND the normal direction
Based on these preparatory studies, the pin references samples from the case-hardened 20MnCr5 steel samples investigated by means of neutron diffraction were sampled by means of EDM. The respective depth distributions of the interplanar distances vs. the sample thickness (corresponds with the pin length) are shown in Figure 6. For the measured hoop direction with regard to the ring geometry, no difference can be found for the two reference samples, i.e., for the pins with a diameter of Ø1.7 mm and Ø 3 mm, respectively. For both pin geometries, a significant increase in the interplanar spacing towards the case-hardened surface can be noticed. A comparison with the interplanar spacings determined in the same way in hoop direction of the solid sample shows that a clear and comparable RS release must have occurred in both pins. Since the results for both pin geometries are comparable, there is no requirement in case of the investigated case-hardened steel samples to use a reference pin with a diameter smaller than 3 mm. This allows selecting the larger pin geometry in future measurements on comparable material conditions, such as hardened gears, which would also permit a larger nominal gauge volume and a much better statistics accompanied with a reduction in measurement time as well as a much more convenient sample alignment.

Results of the neutronographic residual stress analyses on reference pins (\({d}_{0}\) samples). The experimentally determined depth distribution of the interplanar spacing of the reference pins with diameters Ø 1.7 and 3 mm are compared with results determined for the bulk sample in hoop direction (radial direction of the pins corresponds to the hoop direction of the ring—cf. Figures 1 and 3). The dashed line represents a mean distribution for both pins
However, the question arises to which extent the increase of the interplanar spacing towards the end face (hardened surface) of the pin is due to a redistribution of RS or simply due to the chemical composition, i.e., the carbon gradient. In order to answer this question, the lattice strain changes in the martensite phase to be expected due to the carbon gradient are calculated.
Based on analytical approaches described in literature the lattice strains induced by the carbon content in the martensite phase can be calculated. For this purpose, a cell parameter \({a}_{0}\) of 2.866 Å[41] was considered for bcc iron. The deformation of the martensite unit cell represented by the lattice parameters a and c of the tetragonal body centred unit cell can be described as follows[42]:
Here, \(\rho \) denotes the soluted carbon content. The measured values presented in Figure 4 were used for this calculation. The resulting interplanar spacing is calculated according to Reference 43 by:
with h, k, l being the Miller indices. In Figure 7(a) the lattice strain depth distribution as calculated on the base of Eqs. [3] through [5] using the measured carbon distribution as input data are shown. In Figure 7(b) this calculated lattice strain distribution (solid blue line) is compared with experimentally determined lattice strains resulting from the analyses of the two free-cut reference pins. The open triangles in Figure 7(a) represent the distribution calculated according to the measured carbon content (cf. Figure 4). However, regarding the distortion of the crystal lattice it must be taken into account that carbon is also soluted in the retained austenite phase and furthermore contributes to the formation of any carbides that may be present in the structure. This would locally slightly reduce the carbon content in the martensite phase and consequently the lattice strain in martensite that is related to the carbon content. However, this does not explain the observed deviations between the calculated and the experimentally determined lattice strain distributions (cf. Figures 7(a) and (b)). It is obvious that the absolute values of the lattice strain agree quite well. However, it appears that the distributions are slightly shifted along the abscissa, i.e., the sample thickness. This effect might be based on the measurement strategy. While through surface strain scanning a convolution of the lattice strain distribution (resulting from the carbon content) with the gauge volume geometry occurs. Consequently, this convolution is calculated by fitting the carbon influenced lattice strain and describing the nominal gauge volume approximately by means of two-step functions at an interval of 0.6 mm. The resulting lattice strain profile is represented as a solid blue line in Figures 7(a) and (b). The lattice strain distribution calculated in this way indicate that the above mentioned shift along the abscissa is significantly reduced. Thus, it can be assumed that the near-surface increase in interplanar spacing in both pins is primarily due to the carbon-dependent change in the interplanar spacing. However, neglecting the chemical gradient during data evaluation, e.g., by measuring only a location in the bulk of the sample that is expected to be stress-free, would have resulted in a strain error of min. 1200 µstrain (Figures 7 and 8).

(a) Lattice strain depth distribution calculated on the base of Eqs. [3] through [5] using the measured carbon distribution as input data. While through surface strain scanning a convolution of the lattice strain distribution (resulting from the carbon content) with the gauge volume geometry occurs. Accounting for this convolution results in the distribution presented as a solid blue line. In (b) this calculated lattice strain distribution (solid blue line) is compared with experimentally determined lattice strains resulting from the analyses of the two free-cut reference pins. The results show that considering the convolution a better agreement between calculation and experimental data can be obtained. In this figure, the averaged lattice plane spacing from the bulk was taken as the \({d}_{0}\) value (Color figure online)

Correction of pseudo-strains and of the assigned information depth, i.e., the sample thickness due to the partially filled gauge volume for the strain component in radial direction. The grey distribution (open triangles) shows the uncorrected lattice strain if only a constant \({d}_{0}\) from the bulk (mean value radial direction depth between 3 and 6 mm) was used. The black squares (open squares) corresponds to an evaluation with the \({d}_{0}\) reference depth distribution determined for the free-cut pin. In green the resulting distribution is shown, how it results from the STRESSFIT correction for the area near the surface, where the measuring volume is not completely filled, yet. Inside the sample, no correction is necessary. Therefore, the red solid lines distribution is constructed from the black open squares and green filled circles distributions, which is consequently applied in all further calculations (Color figure online)
The still existing deviations between the calculated (solid blue line) and experimentally determined (pin data) lattice strain distributions can be attributed to various effect, as e.g., (i) uncertainties in the assigned reference lattice parameter a0, (ii) the occurrence of phase-specific micro RSs in the near-surface region where significant amount of retained austenite phase is present (cf. Figure 9) and (iii) asymmetries of the martensite interference lines due to the tetragonal distortion of the martensite unit cell by the high carbon content. In this regard, it must be mentioned that the phase-specific micro RSs will not be fully released during free-cutting of the reference pin samples. Here, first of all the long-range macro RSs are released.

Residual stress distribution over the wall thickness of the case-hardened sample, based on the \({d}_{0}\) values determined with the Ø 1.7 mm reference pin. In addition to the neutron diffraction results, the results of lab X-ray stress analysis determined using the sin2ψ-approach at the outer mantle surface is presented. In (a) the residual stress distributions for the ferritic/martensitic phase over the whole sample thickness. For further discussion of the neutron diffraction results, the corresponding hydrostatic stress component is also plotted. In (b) detailed view for the residual stresses of the near-surface outer region. In this partial image, the neutronographic RS were corrected by the amount of the hydrostatic stress component. For the values determined by neutron diffraction, the ferrite/martensite phase was considered only. X-ray stress analysis was also feasible in the retained austenite (RA) phase although the phase fraction was rather small
In the present work for the further discussion, we used the experimentally determined lattice strain distribution resulting from the 1.7 mm pin as a reference, as long as it is not explicitly mentioned otherwise. Due to the explained procedure for the determination of the line position (cf. also Figure 3) in the bulk sample, only the “through-surface-scan” in radial direction results in spurious strains, which require a correction of the line position (pseudo-strains from surface effect) and the associated measurement coordinate. These pseudo-strains can be in the range of several hundred up to a few thousand microstrains. In the present case, using Figure 8, the pseudo-strains can be estimated at about 300 µstrain. At this point, it must be explicitly pointed out that this effect only affects the immediate surface layer until the nominal gauge volume is completely immersed in the sample. A correction of the associated coordinate in this type of immersion scan results from the geometry of the stationary nominal gauge volume, in which the sample is immersed step by step and thus provides information on the position of the diffraction lines exclusively for the entirely immersed volume element. The centre of this immersed volume does not generally coincide with the nominal centre of the nominal gauge volume. A possible mathematical correction approach by hand would be described e.g., in Reference 10 (Appendix 2). A software-supported correction is possible with the simulation tools SIMRES[16,17,18] and STRESSFIT.[19,20] In the other two directions, only a correction of the coordinate according to the centre of gravity of the immersed measuring volume was necessary.
The result of the necessary corrections of the surface effect in radial direction is shown in Figure 8. The distribution with the open triangles shows the non-corrected lattice strain if only a constant reference value \({d}_{0}\) from the bulk (mean value radial direction between 3 and 6 mm), i.e., from a region that is expected to be unstressed, is used. The distribution with the open squares corresponds to an evaluation using the results of the reference measurements performed for the Ø 1.7 mm reference pin. Here, the lattice strain deviations in the near -surface region are obvious, which occur in case the chemical gradient is ignored. Since only the radial direction was averaged for the global constant \({d}_{0}\) in Figure 8, there appears a better agreement in the bulk of the sample than is actually the case. In this sample, the differences in the mean values in the sample core between the three directions are around 0.001 Å. Due to the correction of the sample position (coordinate correction) that has not been carried out yet, it seems that some measuring points are still outside the volume. For the distribution with the open squares, the pseudo-strain correction as well as the correction of the surface position was performed using the software package STRESSFIT. The resulting distribution is represented by the green filled circles distribution in Figure 7. Since the correction was only applied to the region where there is an incompletely filled gauge volume (surface to completely immersed), the final distribution was composed of a combination based on the green filled circles (near-surface region, i.e., not entirely immersed) and the open squares (bulk, i.e., gauge volume entirely immersed) distribution. If the nominal gauge volume is completely filled by sample material, no further correction of the measured data are necessary and hence in case of those bulk values the initial strain distribution (black) is applied. The combined distribution is finally represented by the red solid line distributions, which was also adopted in the further course for the RS calculation according to Eq. [2].
3.3 Residual Stress Distributions
The final RS distributions are presented in Figure 9. In Figure 9(a) the RS distribution over the entire sample thickness is shown. In Figure 9(b) a detailed view with a focus on the outer surface is shown, where the comparison between neutron diffraction and X-ray diffraction (lab method) stress analysis is worked out more clearly. In general, the (phase-specific) RS for the ferrite/martensite phase was determined. The amount of retained austenite phase was larger than 10 vol pct only close to the surface in a depth up to about 0.5 to 0.6 mm due to the carbon depth distribution. This amount is high enough that reasonable RS data could be determined also for the austenite phase by means of X-ray diffraction stress analysis. However, for the neutron diffraction through surface scans the zone, where sufficient amount of retained austenite phase is present, is too localized that no meaningful diffraction data could be recorded for the austenite phase. The X-ray RS analyses in both phases reveal that only small amount of phase-specific RS occur, hence, the RS determined for the martensite/ferrite phase largely represents the macro RS distribution. The difference between phase-specific stresses in ferrite and austenite is around 100 MPa. In the present case and taking into account the phase contents, phase-specific micro stresses arise in the range up to about 75 MPa in the austenite phase and below about 50 MPa in the ferrite phase.
In general it can be stated that by applying the previously describes measuring and evaluation strategy in the core in all three principal directions tensile RS were determined. The lowest amount of tensile RS in the centre (around 4.5 mm) was determined for the radial direction. Towards the outer and the inner surface these tensile RS first increase for all three components. This can be illustrated again for the hoop direction for the results shown in Figure 7(a), provided that the assumption is correct that the pin is almost stress-free. In the region, where the targeted CHD is, i.e., in a distance between 1 and 1.5 mm to the sample surface, the tensile RS in particular for the hoop and the axial direction decrease and compressive RS are build up through LPC processing. At the inner surface ID, the data from neutron stress analyses are poor. This is due to relative low intensities of the recorded diffraction lines due to absorption effects. Close to the inner surface at depths larger than 8 mm no evaluable diffraction information could be determined with the used setup. At the inner surface ID for the RS the general trend can be noticed, but the error bars become rather large. One consequence of this is that an evaluation approach via stress equilibrium is not possible. Since the RS distributions determined at the outer surface are much more trustworthy, the RS induced by LPC processing will be discussed based on the data determined for the outer surface OD. Based on the RS distribution from Figure 9(a), the hydrostatic RS component was calculated in accordance with Reference 44 and this value was subtracted from the neutronographic RS results. The resulting distribution as shown in Figure 9(b), allows for a better comparison of the neutron diffraction data with the RS determined by laboratory XRD analyses. With X-ray diffraction, only deviatoric RS components can be analysed, but neutronographically always the sum of the deviatoric and hydrostatic RS components is determined. By the strict application of the sin2ψ-method from the slope of the regression line 2Θ vs. sin2ψ only the deviatoric stress component is provided. The hydrostatic RS component results in a vertical shift of the regression line, which can only be assessed accurately if an exact value of the local reference value d0 is known. Looking at the overall depth profile on the OD side, there is a pronounced compressive RS plateau up to a depth of about 1.3 mm of about − 115 MPa for the axial component. The tangential RS components drops to about 0 MPa already from a depth of about 0.7 mm on. Furthermore, the radial RS component (Figure 9(a)) shows relatively high tensile RS with a maximum approximately in the CHD region, reaching a value of approx. 400 MPa. Towards the surface, these tensile RS continuously decrease. At a depth of about 0.3 mm, tensile RS of approx. 200 MPa were determined. Since at the very surface the radial stress component must be zero (free surface) it is expected that the tensile RS continuously decrease to zero for smaller depths below the surface. At the OD side, the neutron diffraction results agree rather well with the results determined by means of X-ray diffraction, indicating that the chosen measurement and evaluation strategy for neutron diffraction stress analyses including the correction of the measured date for the surface effect and the chemical gradient is appropriate and reasonable. Furthermore, the RS distributions in hoop and in axial direction close to the surface are in very good agreement with results of earlier work.[25] In this near-surface region, for the austenite phase slightly lower compressive RS of about − 100 MPa were determined. In larger distances to the surface in axial and hoop directions, the compressive RS decrease and balancing tensile RS build up. From the phase-specific RS and the knowledge of the phase content, the phase-specific micro RS can be separated from the macro RS using a simple rule of mixture.[37] The small differences of the RS distribution of about 100 MPa between both phases indicate that rather small phase-specific micro RS (± 50 MPa, depth < 0.8 mm) generate through the LPC process. This is also in agreement with the results from Reference 39. However, for the RS component in radial direction in work that has been done on larger components mostly only small amount of RSs were determined, as e.g., much lower than ± 100 MPa. In the current work largely deviating RS distributions were determined in radial direction.
Regarding these unexpected high RS in radial direction, in particular in the region of the CHD it is assumed that this is supported by the geometry of the component. The geometry of the part in combination with the case-hardening treatment with a CHD of 1.5 mm, which was carried out on the entire surface of the part (none of the sides was masked during the carburizing treatment), is associated with a complex constraint, which can lead to the formation of an locally inhomogeneous RS component, including a relatively high RS component in the direction towards the surface of the part. In the present work, however, the focus was more on a contribution to the measurement and evaluation strategy of such components. Unfortunately, the exact reasons for the RS distributions found cannot be given on the basis of the results presented. Simulation calculations can possibly help here to prove the assumptions. However, due to the complexity of the problem a clear interpretation of the local effects is nearly impossible. First, the resulting triaxial RS distribution is a superposition of quenching and transformation-induced RS. In addition, the RS formation is influenced by the local temperature gradients in all spatial directions of the ring-shape geometry with relative small wall thickness, the transformation kinetics (which again depends on the carbon gradient) and the temporal evolution of the martensite formation during cooling, which is accompanied with a volume expansion. Finally, with the present ring-shaped geometry, the cooling behaviour is also dependent on the quenching intensity, which could also depend on the positioning of the samples relative to the quenching gas stream. However, no specific knowledge is present in case of the here investigated samples (industrial process). Regarding the gas quenching also the local surface-to-volume ratio of the samples can affect the results due to the differences in local heat dissipation. Nevertheless, the RS distributions shown in Figure 8 provide an excellent data basis for validating case-hardening simulations for the purpose of understanding the LPC process of ring-shaped sample geometries. Clear indications of a valid relationship between the geometric shape, the quenching and the RSs that develop in thin annular specimens can be found e.g., in Reference 45.
As a final remark considering the entire RS distribution it appears that no stress equilibrium seem to occur. An excess of tensile RS is visible. However, it should be noted that due to the ring geometry a much larger volume fraction in the outer surface region is actually in compression than the plot vs sample thickness suggests.
4 Conclusions
In the present work, neutronographic RS analysis on LPC case-hardened workpieces was carried out. A special focus was put on the influence of the chemical gradient induced by the carburization process on the RS results and on the choice of the appropriate measuring and evaluation strategy. The following conclusions can be drawn from the investigations:
-
The results indicate that the chemical gradient induced by the carburization step must be taken into account for RS evaluation. Neglecting this gradient leads to quite erroneous results. According to Figure 7, an error of approximately 1500 µstrain in the determination would have been made in this work when determining \({d}_{0}\) if the chemical gradient had not been taken into account. In this case, this would result in an approximate error of up to about 750 MPa.
-
The depth distribution of the stress-free reference values \({d}_{0}\) must be determined for a stress relaxed reference sample sectioned from a region with a similar microstructure region, possibly from a twin sample. Those results must be applied for RS evaluation.
-
Alternatively, for case-hardened steel samples the required \({d}_{0}\) depths distribution can be calculated based on the known carbon content depth distribution and the measurement of one \({d}_{0}\) value from the bulk as support point for the calculation. Due to the nature of the neutron diffraction experiment, i.e., integrating over a distinct nominal gauge volume, a convolution of the carbon depth distribution with the nominal gauge volume dimension must be considered in this regard. As limitation it must be mentioned, that this only applies to the quenched and non-tempered state as investigated in the present study.
-
For the present example of a ring-shaped LPC case-hardened steel sample a cylindrical reference pin processed by means of EDM with a diameter of 3 mm is sufficiently small that the macro RS can relax through the sectioning.
-
Since sectioning induces RS in a very shallow surface region of the reference sample, this region can be considered by ensuring that the reference sample size is slightly larger than the nominal gauge volume dimension. In the present case (EDM cutting) the sample should be at least 200 µm larger in diameter than the diagonal of the neutronographic gauge volume.
-
The strategy for correcting the surface effects induced by neutron through surface scanning worked out for single-phase gradient-free materials in previous work was successfully transferred to materials with chemical gradients. The application of the mathematical correction using the freely available programs SIMRES and STRESSFIT on the LPC processed sample showed an effective correction of the pseudo-strains.
-
In the directions parallel to the surface, the low-pressure carburization process-induced compressive macro RS of about − 100 to − 200 MPa close to the outer surface of the ring sample. The rather high amount of retained austenite phase in the near-surface region only contributes to very low amount of phase-specific micro RS.
-
In depths beyond about 0.8 mm the compressive RS markedly decrease and the near-surface compressive RS are balanced by tensile RS in the bulk.
-
From a depth of about 0.2 mm, reliable RS results can be determined by neutron diffraction. In combination with results from X-ray diffraction stress analysis from the region close to the surface, enables the complete description of the RS distribution over the entire component thickness.
References
V. Argoud, F. Morel, E. Pessard, D. Bellett, S. Thibault, and S. Gourdin: Procedia Struct. Integr., 2019, vol. 19, pp. 719–28.
P. Wei, H. Zhou, H. Liu, C. Zhu, W. Wang, and G. Deng: Int. J. Mech. Sci., 2019, vol. 156, pp. 283–96.
D.C. Goss: J. Mater. Process. Technol., 2000, vol. 98, pp. 135–42.
S. Němeček, M. Míšek, I. Černý, J. Sís, N. Ganev, and K. Kolařík: MSF, 2014, vol. 782, pp. 306–10.
M. Steinbacher: Thermogravimetrische Messungen beim Niederdruckaufkohlen als Grundlage für Simulationen, 2012. http://nbn-resolving.de/urn:nbn:de:gbv:46-00102752-13. Accessed 9 Dec 2022.
S. Bischoff, H. Klümper-Westkamp, F. Hoffmann, H.-W. Zoch, and J. Rohde: HTM J. Heat Treat. Mater., 2013, vol. 68, pp. 199–206.
E28 Committee: Test Method for Determining Residual Stresses by the Hole-Drilling Strain-Gage Method, ASTM International, West Conshohocken.
T. Erbacher, A. Wanner, T. Beck, and O. Vöhringer: J. Appl. Crystallogr., 2008, vol. 41, pp. 377–85.
L. Spieß: Moderne Röntgenbeugung: Röntgendiffraktometrie für Materialwissenschaftler, Physiker und Chemiker, 2nd ed. Vieweg+Teubner, Wiesbaden, 2009.
M.T. Hutchings, P.J. Withers, T.M. Holden, and T. Lorentzen: Introduction to the Characterization of Residual Stress by Neutron Diffraction, 1st ed. CRC Press Taylor & Francis Group, Boca Raton, 2005.
P.J. Withers: J. Appl. Crystallogr., 2004, vol. 37, pp. 596–606.
T. Lorentzen: J. Neutron Res., 1997, vol. 5, pp. 167–80.
X.-L. Wang, S. Spooner, and C.R. Hubbard: J. Appl. Crystallogr., 1998, vol. 31, pp. 52–59.
T. Pirling: MSF, 2000, vol. 347–349, pp. 107–12.
T. Pirling, D.J. Hughes, and J.S. Robinson: MSF, 2010, vol. 652, pp. 80–85.
J. Šaroun and J. Kulda: SIMRES: Monte Carlo Neutron Ray-Tracing Simulations, 2020. http://neutron.ujf.cas.cz/restrax/. Accessed 9 Dec 2022.
J.R. Kornmeier, J. Šaroun, J. Gibmeier, and M. Hofmann: MSF, 2013, vol. 768–769, pp. 52–59.
J. Šaroun and J. Kulda: in Modern Developments in X-Ray and Neutron Optics. A. Erko, M. Idir, T. Krist, and A.G. Michette, eds., Springer, Berlin, 2008, pp. 57–68.
J. Šaroun, J. Rebelo-Kornmeier, J. Gibmeier, and M. Hofmann: Physica B, 2018, vol. 551, pp. 468–71.
J. Šaroun: STRESSFIT—Fitting Of Residual Stress Distributions, 2020. https://github.com/NPLtools/stressfit. Accessed 2021.
S. Pulvermacher, T. Bücker, J. Šaroun, J. Rebelo-Kornmeier, M. Hofmann, and J. Gibmeier: Materials (Basel Switz.), 2021, vol. 14, p. 1854.
P.J. Withers, M. Preuss, A. Steuwer, and J.W.L. Pang: J. Appl. Crystallogr., 2007, vol. 40, pp. 891–904.
J. Epp, T. Hirsch, M. Hunkel, and R.C. Wimpory: MSF, 2010, vol. 652, pp. 37–43.
J. Epp, T. Pirling, and T. Hirsch: MSF, 2013, vol. 768–769, pp. 420–27.
J. Gibmeier, J. Rebelo-Kornmeier, and T. Strauss: MSF, 2016, vol. 879, pp. 601–06.
N. Bordel: Anal. Bioanal. Chem., 2005, vol. 382, pp. 863–64.
Z. Zhou, K. Zhou, X. Hou, and H. Luo: Appl. Spectrosc. Rev., 2005, vol. 40, pp. 165–85.
E04 Committee: Practice for X-Ray Determination of Retained Austenite in Steel with Near Random Crystallographic Orientation, ASTM International, West Conshohocken.
DIN EN ISO 14577-1:2015, Beuth Verlag GmbH, 2015-11-00, vol. 77.040.10.
DIN EN ISO 14577-2:2015, Beuth Verlag GmbH, 2015-11-00, vol. 77.040.10.
DIN EN ISO 14577-3:2015, Beuth Verlag GmbH, 2015-11-00, vol. 77.040.10.
DIN EN ISO 14577-4:2016, Beuth Verlag GmbH, 2015-11-00, vol. 77.040.10.
J. Gibmeier, S. Cabeza, M. Hofmann, T. Pirling, S. Pulvermacher, J. Rebelo Kornmeier, and J. Saroun: Residual Stress Analysis for Materials with Depth Gradients of the Strain Free/Independent Lattice Parameter d0, 2019. https://doi.org/10.5291/ILL-DATA.1-02-277.
D. Lober: Informationen über Stahl für Metallografen - Klemm-I Ätzmittel (21.05.2018). www.metallograf.de. Accessed 2021.
T. Pirling, G. Bruno, and P.J. Withers: Mater. Sci. Eng. A, 2006, vol. 437, pp. 139–44.
D. Richard, M. Ferrand, and G.J. Kearley: J. Neutron Res., 1996, vol. 4, pp. 33–39.
B. Eigenmann and E. Macherauch: Mat.-wiss. Werkst., 1995, vol. 26, pp. 148–60.
Landolt-Börnstein: Neue Serie, Springer, Berlin, 1966.
M.G. Zuern, O.B. Tapar, P. Ho, J. Epp, and J. Gibmeier: HTM J. Heat Treat. Mater., 2022, vol. 77, pp. 29–52.
DIN EN ISO 2639:2002, Beuth Verlag GmbH, 2003-04-00, vol. 77.040.99.
K.-H. Hellwege and A.M. Hellwege: Structure Data of Elements and Intermetallic Phases, Springer, Berlin, 1971.
L. Cheng, A. Böttger, T.H. de Keijser, and E.J. Mittemeijer: Scripta Metall. Mater., 1990, vol. 24, pp. 509–14.
Y. Chen, W. Xiao, K. Jiao, D. Ping, H. Xu, X. Zhao, and Y. Wang: Phys. Rev. Mater., 2018, vol. 2, p. 25.
W. Soboyejo and W.O. Soboyejo: Mechanical Properties of Engineered Materials, Dekker, New York, 2003.
R. Kübler: Numerische und experimentelle Untersuchungen zum Einfluß des Wärmeübergangs beim Hochdruckgasabschrecken in Düsenfeldern auf die Spannungs-, Eigenspannungs- und Verzugsausbildung von rotationssymmetrischen Proben aus 100 Cr 6. Zugl.: Karlsruhe University, Dissertation, 2001, Shaker, Aachen.
Acknowledgments
The authors would also like to thank Mr. Taper from IWT-Bremen for the analysis of the near-surface carbon and manganese gradients. In addition, we would like to thank the Institute-Laue-Langevin, Grenoble (France) for providing beam time 1-02-277 [33] at the SALSA instrument at the neutron research reactor.
Author Contributions
Conceptualization, SP and JG; methodology, SP and JG; software, SP and JŠ; validation, SP, JŠ and JG; formal analysis, SP; investigation, SP, SC and TP; resources, MH and JG; data curation, SP; writing—original draft preparation, SP; writing—review and editing, SP, SC, TP, JS, JE, JR-K, MH and JG; visualization, SP; supervision, MH and JG; project administration, MH and JG; funding acquisition, MH and JG. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the German Research Foundation (DFG) under the Grant Numbers Gi376/11-1, Ho 3322/4-1 and the Czech Science Foundation GACR for financial support through the Project No. 16-08803J.
Data Availability
The data presented in this study are available on request from the corresponding author.
Conflict of interest
The authors declare no conflict of interest.
Ethical Approval
Not applicable.
Informed Consent
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pulvermacher, S., Šaroun, J., Cabeza, S. et al. Neutronographic Residual Stress Analysis for Materials With Depth Gradients of the Strain Free Lattice Parameter \({{\varvec{d}}}_{0}\) for the Example of a Case-Hardened Steel 20MnCr5. Metall Mater Trans A 54, 3448–3460 (2023). https://doi.org/10.1007/s11661-023-07076-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-023-07076-y