
Abstract
The total arsenic mass fraction as well as the arsenic speciation were studied in four different mushroom species with inductively coupled plasma mass spectrometry and high-performance liquid chromatography coupled to inductively coupled plasma mass spectrometry, respectively. Arsenic mass fractions detected in the mushrooms were covering a range from 0.3 to 22 mg As kg−1 dry mass. For the arsenic speciation, species like arsenobetaine, inorganic arsenic, or dimethylarsinic acid were found, which are commonly detected in mushrooms, but it was also proven that the recently discovered novel compound homoarsenocholine is present in Amanita muscaria and Ramaria sanguinea. Moreover, a previously unidentified arsenic species was isolated from Ramaria sanguinea and identified as trimethylarsonioacetamide, or in short: arsenobetaine amide. This new arsenical was synthesized and verified by spiking experiments to be present in all investigated mushroom samples. Arsenobetaine amide could be an important intermediate to further elucidate the biotransformation pathways of arsenic in the environment.
Graphical Abstract

Similar content being viewed by others

Introduction
In a study about the global fungal diversity from 2021, including published datasets of roughly a million different fungal species, the total number of fungal species present in the world was estimated to be over six million [1]. Some of these fungi, specifically mushrooms, are known to have an extremely diverse arsenic speciation in their fruiting-bodies. In contrast to other organisms, it is almost impossible to predict the arsenic speciation in mushrooms just by comparison with similar mushroom species [2,3,4]. Some mushrooms contain mainly inorganic arsenic (iAs, in the form of arsenate and arsenite), dimethylarsinic acid (DMA), and/or methylarsonic acid (MA) (Fig. 1) which are usually found in terrestrial organisms [5,6,7]. However, the most often found main arsenic species in mushrooms is arsenobetaine (AB), which is commonly found in marine animals [8]. Furthermore, traces of arsenocholine (AC), the tetramethylarsonium ion (TETRA), trimethylarsine oxide (TMAO) (Fig. 1), and unidentified arsenic species can be detected in mushrooms and sometimes even as the main species [3, 4, 7, 9, 10].

Arsenic species relevant for this publication
It is still unclear whether mushrooms are able to metabolize arsenic or if they simply take up different arsenic species from the substrate or if other microorganisms are involved in these processes. A number of studies tried to investigate this question, but no definitive answer could be found [11, 12]. To further elucidate the pathway of arsenic and its possible biotransformation, identifying intermediate compounds could be of valuable help. Therefore, it is important to identify previously unknown arsenic species [10, 13, 14], which will hopefully help us to better understand the reason behind the diversity of arsenic species in mushrooms.
In a previous study on edible mushrooms, Boletus edulis (king bolete) and Macrolepiota procera (parasol mushroom) proved to exhibit a complex distribution of arsenic species, including some hitherto not identified arsenic species [15]. Other good candidates for interrogating potential intermediates would be the famous Amanita muscaria (fly agaric), which was already reported to contain several unidentified arsenic species [4, 9], as well as the Ramaria genus, since they also possess a complex arsenic speciation profile and tend to take up more arsenic than any of the mushrooms mentioned above [10]. This higher mass fraction of arsenic would simplify the process of isolating a potentially new arsenic compound and identifying it.
In this study, we investigated the arsenic speciation in B. edulis, M. procera, A. muscaria, and Ramaria sanguinea. We employed high-performance liquid chromatography (HPLC) coupled to inductively coupled plasma mass spectrometry (ICPMS) to unravel the arsenic speciation of these mushrooms with some unidentified signals and aimed to identify one of the unknown arsenic species. Therefore, we thoroughly enriched and purified the unknown compound by collecting fractions of mushroom extracts by HPLC and freeze-drying the pooled collection. Finally, we identified a novel nitrogen-containing arsenic species using HPLC coupled to high-resolution mass spectrometry. This new arsenic compound will give us a new perspective in the current understanding of the arsenic speciation in the environment and add to the knowledge about the biotransformation ways of arsenic.
Materials and methods
Mushroom collection and processing
For this study, we used eleven individual mushroom fruiting-bodies collections as samples, which were collected in different regions of Austria and the Czech Republic. From three of the samples (B. edulis 1 + 4 and M. procera 1), the arsenic speciation was already published and discussed in a previous publication [15]. We investigated mushroom samples from non-contaminated as well as arsenic-contaminated regions (summarized in Supplementary Information Table S1). These samples were selected, because preliminary analysis of these mushroom species showed them to have a very diverse arsenic speciation profile and many unknown arsenic species with similar chromatographic behavior. The sample handling, sample preparation, and determination of the total arsenic content as well as the determination of the common water-soluble arsenic species are described in detail elsewhere [15].
Briefly, the samples were directly brought to the laboratory, where they were carefully cleaned before freeze-drying (Christ, Osterode am Harz, Germany) and homogenization (ultra-centrifugal mill ZM200, 1 mm titanium sieve, Retsch GmbH, Haan, Germany).
The freeze-dried mushrooms were digested in triplicates with nitric acid (p.a., ≥65%; Carl Roth, Karlsruhe Germany, sub-boiled in-house) in a microwave-heated autoclave (Ultraclave IV, MLS GmbH, Leutkirch, Germany; temperature ramp up to 250 °C, pressure up to around 100 bar) and investigated with ICPMS (7700x, Agilent Technologies, Waldbronn, Germany) for the determination of the total arsenic mass fraction. The performance of the instrument can be found in Table S2. For quality control, the Standard Reference Material® (SRM) 1573a (Tomato Leaves, NIST, Gaithersburg, USA), the certified reference material (CRM) IPE-120 (Agaricus bisporus, WEPAL, Wageningen, Netherlands), and the SRM® 1643f (Trace elements in water, NIST, Gaithersburg, USA) as well as blanks consisting of ultrapure water (18.2 MΩ*cm, Merck Millipore, Bedford, USA) were prepared and measured with the samples. To control the stability of the measurement, germanium was added as internal standard via a t-piece before the nebulizer.
For arsenic speciation analysis, dried mushroom samples were extracted in triplicates with ultrapure water and investigated by coupling HPLC (1200, Agilent Technologies) to ICPMS with carbon dioxide as optional gas, added via optional gas line, to compensate for the carbon enhancement effect. An anion-exchange column (PRP-X100) with an aqueous 20 mM phosphate buffer (pH 6.0) and a cation-exchange column (Reprosil-XR 300 SCX) with an aqueous 10 mM pyridine solution (pH 2.3) were applied to detect and quantify iAs, DMA, MA, AB, TMAO, AC, and TETRA using an external calibration containing these arsenic species. The results for the quality control of all measurements were in good accordance with the certified or published data [6, 13, 15] (Table S3 and S4). The total arsenic mass fraction of the extracts was determined by diluting the samples 1+9 with 10% v/v nitric acid and directly measuring it with ICPMS.
A quadrupole time of flight MS (6546, Agilent Technologies) was used for high-resolution electrospray ionization mass spectrometry (HR ESI-MS) measurements in positive mode (Table S5). It was coupled to HPLC equipped with a cation-exchange column (Zorbax 300-SCX, 4.6 × 150 mm, 5 µm, Agilent Technologies) at 30 °C, 30 mM ammonium formate, pH 5.0 as mobile phase, and a flow rate of 0.6 mL min−1.
All 1H and 13C NMR spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) on an Avance III 300 MHz spectrometer (Bruker Corporation, Billerica, USA) at 300 K and the spectra were processed using the MestreNova software (Mestrelab Research).
Results and discussion
The total arsenic mass fraction in the different mushroom samples ranged from 0.28 ± 0.01 mg kg−1 dry mass (dm) found in a B. edulis sample to 22 ± 2 mg kg−1 dm found in an A. muscaria sample (Table 1). The extraction efficiency was determined by comparing the total arsenic mass fraction found after digestion with nitric acid, with the total arsenic mass fraction found in the aqueous extracts. Across all the analyzed mushrooms, the extraction efficiency was 77 ± 11 % extracting the water-soluble arsenicals. For column recovery, the cation-exchange chromatogram was integrated at the baseline over the whole time span and quantified via compound independent calibration using AB for calibration. This arsenic mass fraction recovered in the chromatogram was compared to the total arsenic mass fraction found directly in the extracts to give a column recovery of 99 ± 6 %.
AB was the main arsenic species found in all mushroom samples, except the sample B. edulis 1, which had DMA as the main arsenic species. However, this mushroom species is known to contain some unidentified arsenicals and has no clear trend to a certain arsenic species [15]. We also found different amounts of iAs, MA, TMAO, TETRA, trimethylarsoniopropionic acid (AB2), and homoarsenocholine (AC2) (Fig. 1). These arsenic species were confirmed by spiking and co-chromatography. The most important results are given in Table 1. Mass fractions of all detected arsenic species can be found in Table S6. As a side note, we now proved, as already speculated in a previous report, that AC2 is indeed present in A. muscaria and can also be detected in the R. sanguinea [10].
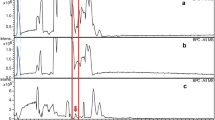
Several unassigned peaks were found, especially in the cation-exchange chromatograms (see Fig. 2). With spiking experiments, we excluded dimethylarsinoylbutanoic acid (DMAB) and trimethylarsoniobutanoic acid (TMAB) as possible candidates (Fig. 1). Furthermore, addition of hydrogen peroxide to the sample proved that no known thio-arsenic compound was present, and that the unknown compounds likely contained arsenic in oxidation state +5.

Typical anion-exchange (a) and cation-exchange (b) chromatogram of a Macrolepiota procera 2 extract and a cation-exchange chromatogram (c) of a Ramaria sanguinea extract. Dotted lines show the extracts spiked with synthetic AB-amide. The small arrow indicates the void time of the method used
Our attention was mainly attracted by unknown compound UNK 1, eluting between TMAO and AB2, because it was present in detectable amounts in all investigated samples. Its mass fraction was highest in R. sanguinea, accounting for 1.65 ± 0.01 mg As kg−1 dm. This promised a good possibility to clarify the structure of UNK 1.
To further enrich the concentration of UNK 1 in solution, we adapted the cation-exchange method to an ammonium formate buffer instead of pyridine, which would sublime via freeze-drying. Fractions mainly containing UNK 1 were obtained with this adapted method by injecting an aqueous extract of the mushroom (R. sanguinea) multiple times onto the cation-exchange column and collecting the effluent around the retention time of UNK 1.
After the collection, the fractions were pooled, frozen, and freeze-dried, before the residue was redissolved in a small volume of ultrapure water. The presence of UNK 1 in the isolate was confirmed with HPLC-ICPMS and spiking it to the R. sanguinea extract. By introducing the isolate to HR ESI-MS, we detected at the corresponding elution time, as specified with HPLC-ICPMS, a molecule with an exact mass of m/z of 178.0207 (Δ= −0.2 ppm) and a proposed sum formula of C5H13AsNO+. Characteristic fragments of the compound under fragmentation experiments can be seen in Fig. 3.

Mass spectrum of HR ESI-MS of the UNK 1 (AB-amide) fraction collected
The m/z 161 represents an ammonia loss, m/z 137 corresponds to the loss of vinylamine, m/z 120 is a charged Me3As, m/z 103/105 is Me2As-2H and Me2As, and m/z 75 is the charged As. Similar fragments can be found for the fragmentation of other arsenicals [16], but the nitrogen group’s exact location in the molecule could not be determined with the obtained HR ESI-MS data. We assumed that it would be preferably at the same carbon as the oxygen and could possibly be a derivate of AB, so we used an adapted literature procedure for AB [17], to synthesize trimethylarsonioacetamide, which we called arsenobetaine amide (“AB-amide”, Fig. 1).
In a nitrogen flushed Schlenk flask equipped with a stirring bar, trimethylarsine (1 mL, 9.4 mmol) was added to 10 mL of dry, deoxygenated toluene. The flask was cooled to 0 °C in an ice bath. 2-Bromoacetamide (1.38 g, 7.2 mmol) was added to the solution and the mixture was stirred at room temperature for 48 h. The formed solids were collected by filtration in air and washed with acetone to afford 473 mg (25 %) of AB-amide as colorless crystals. The purity and structure were confirmed with 1H and 13C NMR experiments and HR ES-MS (see Fig. 4).
-
1H NMR (300 MHz, DMSO-d6): δ = 7.91 (s, NH, 1H), 7.48 (s, NH, 1H), 3.58 (s, CH2, 2H), 1.90 (s, 3 x Me, 9H);
-
13C NMR (75 MHz, DMSO-d6): δ = 167.9, 32.9, 8.7.

Mass spectrum of HR ESI-MS of the synthesized AB-amide and potential structures [16] of the fragments detected
The results were in accordance with our findings of UNK 1, and a successful spiking of the standard to all mushroom samples analyzed further supported that UNK 1 is indeed AB-amide.
To our knowledge, the synthesis of AB-amide has not been published as of now, and the molecule was never reported in any previous study. Additionally, even the analogous ammonium-compound betaine-amide itself is hardly ever explored in scientific literature [18]. That these compounds have almost never been studied or mentioned before is particularly interesting, especially since their chemical structures are so similar to better-studied derivatives such as the arsenic-containing AB [19,20,21] and the nitrogen analogue glycine betaine [21, 22].
Currently there are different pathways proposed how AB is synthesized in marine or terrestrial environments [20, 23, 24]. For most of them, the final biotransformation step to achieve AB is the oxidation of AC or the methylation of dimethylarsinous acid. As AB-amide is a derivate of AB, it is possible that the amide could fit into previously reported biotransformation pathways of AB as a stable intermediate step, as a side reaction product or that AB-amide is a result of a different starting or intermediate compound. This last option would imply that other nitrogen-containing arsenic species such as the amine analogue of AC or the amide analogue of DMAA may also be present. A clear proof to explain how AB-amide fits into the whole biotransformation of arsenic in these mushrooms is not existing and further experiments will be needed for clarification.
In general, reports of nitrogen-containing arsenic species are rarely seen. Historically, some nitrogen-containing arsenic species were developed and applied as drugs, such as arsanilic acid (Atoxyl) against sleeping sickness (trypanosomiasis) and arsphenamine, better known as Salvarsan, as treatment of syphilis [25, 26]. To our knowledge, in natural samples, only the nitrogen-containing arsinothricin [27], which is produced by a rice bacterium, and a handful of arsenosugars with nitrogen-containing side groups were previously identified [28, 29].
Conclusion and outlook
In this report, AB-amide, a nitrogen-containing arsenic species, was identified for the first time in a natural sample. In the investigated mushrooms, AB-amide accounted for 0.15 to 9.6% of total As or in mass fractions 0.002 to 1.7 mg As kg−1 dm. This discovery sheds a new light on the understanding of the biogeochemical pathway of arsenic in the environment. Further identification of other arsenic species in the environment, especially nitrogen-containing ones, would help to better understand the potential role of AB-amide. Looking at chromatograms of previous publications, it can be speculated that “unknown” arsenic species in mushrooms [4, 10] and potentially even in sea snails [30] could indeed be AB-amide. In the future, additional experiments are needed to prove this claim, but if it is verified that AB-amide is present in sea snails, this compound would not only play a role in the biotransformation pathway of arsenic in the fungal kingdom but also in others. This highlights the importance of identifying new arsenic compounds even more to complete our knowledge of the arsenic biotransformation pathways.
Data availability
The data that support this study is shared in the supplementary material. Further data will be shared upon reasonable request to the corresponding author.
References
Baldrian P, Větrovský T, Lepinay C, Kohout P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers. 2022. https://doi.org/10.1007/s13225-021-00472-y.
Braeuer S, Goessler W. Arsenic species in mushrooms, with a focus on analytical methods for their determination - A critical review. Anal Chim Acta. 2019. https://doi.org/10.1016/j.aca.2019.04.004.
Nearing MM, Koch I, Reimer KJ. Arsenic speciation in edible mushrooms. Environ Sci Technol. 2014. https://doi.org/10.1021/es5038468.
Šlejkovec Z, Byrne AR, Stijve T, Goessler W, Irgolic KJ. Arsenic Compounds in Higher Fungi. Appl Organomet Chem. 1997. https://doi.org/10.1002/(SICI)1099-0739(199708)11:8%3c673::AID-AOC620%3e3.0.CO;2-1.
Slekovec M, Goessler W, Irgolic KJ. Inorganic and organic arsenic compounds in Slovenian mushrooms: comparison of arsenic-specific detectors for liquid chromatography. Chem Speciation Bioavailability. 1999. https://doi.org/10.3184/095422999782775618.
Llorente-Mirandes T, Barbero M, Rubio R, López-Sánchez JF. Occurrence of inorganic arsenic in edible Shiitake (Lentinula edodes) products. Food Chem. 2014. https://doi.org/10.1016/j.foodchem.2014.02.081.
Braeuer S, Borovička J, Goessler W. A unique arsenic speciation profile in Elaphomyces spp. (“deer truffles”)-trimethylarsine oxide and methylarsonous acid as significant arsenic compounds. Anal Bioanal Chem. 2018;410:2283–90. https://doi.org/10.1007/s00216-018-0903-3.
Molin M, Ulven SM, Meltzer HM, Alexander J. Arsenic in the human food chain, biotransformation and toxicology–Review focusing on seafood arsenic. J Trace Elem Med Biol. 2015. https://doi.org/10.1016/j.jtemb.2015.01.010.
Kuehnelt D, Goessler W, Irgolic KJ. Arsenic Compounds in Terrestrial Organisms II: Arsenocholine in the Mushroom Amanita muscaria. Appl Organomet Chem 1997; https://doi.org/10.1002/(SICI)1099-0739(199706)11:6<459::AID-AOC583>3.0.CO;2-O
Braeuer S, Borovička J, Glasnov T, de la Cruz Guedes G, Jensen KB, Goessler W. Homoarsenocholine - A novel arsenic compound detected for the first time in nature. Talanta. 2018;188:107–10. https://doi.org/10.1016/j.talanta.2018.05.065.
Šlejkovec Z, Byrne AR, Goessler W, Kuehnelt D, Irgolic KJ, Pohleven F. Methylation of arsenic in Pleurotus sp. and Agaricus placomyces. Acta Chim Slov. 1996;43:269–83.
Nearing MM, Koch I, Reimer KJ. Uptake and transformation of arsenic during the vegetative life stage of terrestrial fungi. Environ Pollut. 2015. https://doi.org/10.1016/j.envpol.2014.12.006.
Braeuer S, Borovička J, Glabonjat RA, Steiner L, Goessler W. Arsenocholine-O-sulfate: A novel compound as major arsenic species in the parasitic mushroom Tolypocladium ophioglossoides. Chemosphere. 2021. https://doi.org/10.1016/j.chemosphere.2020.128886.
Raab A, Kubachka K, Strohmaier M, Preihs M, Feldmann J. New arsenic compound identified in rice grain: dimethylarsonyldimethylarsinic acid. Environ Chem. 2022. https://doi.org/10.1071/EN22063.
Walenta M, Braeuer S, Goessler W. Arsenic speciation of commonly eaten mushrooms from central Europe. Environ Chem. 2023. https://doi.org/10.1071/EN22069.
McSheehy S, Guo X-M, Sturgeon RE, Mester Z. Photochemical alkylation of inorganic arsenic: part 2. Identification of aqueous phase organoarsenic species using multidimensional liquid chromatography and electrospray mass spectrometry. J Anal At Spectrom. 2005. https://doi.org/10.1039/b503662c.
Minhas R, Forsyth DS, Dawson B. Synthesis and characterization of arsenobetaine and arsenocholine derivatives. Appl Organomet Chem. 1998. https://doi.org/10.1002/(SICI)1099-0739(199808/09)12:8/9%3c635::AID-AOC772%3e3.0.CO;2-J.
Tabisz Ł, Pankiewicz R, Rozwadowski Z, Łęska B. Hybrid materials comprising trimethylglycinamide groups: immobilization consequences for anion binding affinities. Tetrahedron. 2015. https://doi.org/10.1016/j.tet.2015.02.056.
Popowich A, Zhang Q, Le XC. Arsenobetaine: the ongoing mystery. Natl Sci Rev. 2016. https://doi.org/10.1093/nsr/nww061.
Caumette G, Koch I, Reimer KJ. Arsenobetaine formation in plankton: a review of studies at the base of the aquatic food chain. J Environ Monit. 2012. https://doi.org/10.1039/c2em30572k.
Stiboller M, Raber G, Francesconi KA. Simultaneous determination of glycine betaine and arsenobetaine in biological samples by HPLC/ICPMS/ESMS and the application to some marine and freshwater fish samples. Microchem J. 2015. https://doi.org/10.1016/j.microc.2015.04.022.
Kidd MT, Ferket PR, Garlich JD. Nutritional and osmoregulatory functions of betaine. World’s Poul Sci J. 1997. https://doi.org/10.1079/WPS19970013.
Zhang W, Miao A-J, Wang N-X, Li C, Sha J, Jia J, Alessi DS, Yan B, Ok YS. Arsenic bioaccumulation and biotransformation in aquatic organisms. Environ Int. 2022. https://doi.org/10.1016/j.envint.2022.107221.
Rahman MA, Hassler C. Is arsenic biotransformation a detoxification mechanism for microorganisms? Aquat Toxicol. 2014. https://doi.org/10.1016/j.aquatox.2013.11.009.
Paul NP, Galván AE, Yoshinaga-Sakurai K, Rosen BP, Yoshinaga M. Arsenic in medicine: past, present and future. Biometals. 2023. https://doi.org/10.1007/s10534-022-00371-y.
Lykknes A, Kvittingen L. Arsenic: Not So Evil After All? J Chem Educ. 2003. https://doi.org/10.1021/ed080p497.
Kuramata M, Sakakibara F, Kataoka R, Yamazaki K, Baba K, Ishizaka M, Hiradate S, Kamo T, Ishikawa S. Arsinothricin, a novel organoarsenic species produced by a rice rhizosphere bacterium. Environ Chem. 2016. https://doi.org/10.1071/EN14247.
Francesconi K, Kuehnelt D. Arsenic compounds in the environment. Environmental Chemistry of Arsenic. New York: Marcel Dekker Inc.; 2002.
Francesconi KA, Edmonds JS, Stick RV. Arsenic compounds from the kidney of the giant clam Tridacna maxima: isolation and identification of an arsenic-containing nucleoside. J Chem Soc Perkin Trans 1. 1992; https://doi.org/10.1039/P19920001349
Maher W, Foster S, Krikowa F. Arsenic species in Australian temperate marine food chains. Mar Freshwater Res. 2009. https://doi.org/10.1071/MF08256.
Acknowledgements
The authors want to thank Panagiotis Bizirtsakis, Jaqueline Rieger, and Katharina Gingl for their support in the lab. Also, thanks to Prof. Mösch-Zanetti for making laboratory space available during the synthesis. The authors also want to thank the organisation ‘Arbeitskreis heimische Pilze’ (Universalmuseum Joanneum) for their help in the sample collection. The work of J.B. was supported by Long-term Development Projects RVO67985831 and RVO61389005. The authors acknowledge the financial support by the University of Graz.
Funding
Open access funding provided by University of Graz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Parts of this work were presented at the ASAC Young Chemists Forum 2023 in Leoben, Austria, and have been awarded with an ABC Best Lecture Prize.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walenta, M., Raab, A., Braeuer, S. et al. Arsenobetaine amide: a novel arsenic species detected in several mushroom species. Anal Bioanal Chem 416, 1399–1405 (2024). https://doi.org/10.1007/s00216-024-05132-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-024-05132-z